The development of massively parallel sequencing has fueled genome-centric cancer research. This technology has enabled us to sequence entire cancer genomes and transcriptomes in a relatively short period of time. However, the data produced by this approach is highly complex and difficult to analyze in order to extract the biologically relevant information. Within our group we are developing new computational methods and analyze sequencing data from tumor specimens.
Sequencing of entire cancer genomes provides a comprehensive portrait of the genomic alterations that have occurred during tumorigenesis. The most common changes are subtle sequence alterations (simple substitutions, small insertions and deletions) followed by structural changes such as copy number alterations and genomic rearrangements. On a genome-wide scale, the number of all these alterations is typically within the range of 104–106events and varies significantly between tumor types with pediatric tumors at lower end of the spectrum and lung cancers, melanoma is being the most mutated cancer genomes. However, most of these mutations are likely to have no functional relevance to the cancer cells. Thus, only a minority of mutations is biologically relevant and can be associated to the pathogenesis of the cancer cells. Finding these genome alterations from sequencing data of large collectives of patient samples is still a challenging task.
Our computational methods played a central role in the analysis of 110 whole small cell lung cancer (SCLC) genomes together with 81 whole transcriptomes and 149 Affymetrix 6.0 SNP arrays for copy number analysis. We obtained a first comprehensive landscape of small cell lung cancer by applying our computational approaches to assess recurrent events in point mutations, copy numbers, and rearrangements5. In particular, we found an almost universal bi-allelic deactivation of TP53 and RB1 in these tumors and can therefore be considered as hallmark of small cell lung cancer. By clustering genomic rearrangements, we were able to detect an accumulation of small translocations within TP73. These translocations leaded to a skipping of several exons, which was confirmed by an integrative genome, transcriptome analysis. In fact, these variants of TP73 have be described earlier to act dominant negative on wild type p53 and p73 and could therefore be considered as oncogene in small cell lung cancer. Furthermore, we found a significant enrichment of damaging mutations in NOTCH family genes. This finding points towards a tumor suppressive function of NOTCH in small cell lung cancer. Indeed, overexpressing NOTCH2 in a Trp53;Rb1;Rbl2 conditional triple-knockout mouse model confirmed this notion as substantially less tumors and a prolonged tumor development was observed.
By analyzing the genomes of 56 primary neuroblastomas we discovered genomic rearrangements affecting a region upstream the telomerase reverse transcriptase gene (TERT) in more than 20% of high-risk tumors4. These rearrangements occurred mutually exclusive with MYCN amplifications and ATRX mutations in most of the cases. Moreover, a more than 90-fold increase of TERT expression in the presence of the rearrangement was observed when compared to low-risk tumors. Since MYCNtranscriptionally activates TERT, high TERT expression and the presence of telomerase activity was observed in tumors bearing TERT rearrangements and MYCN amplifications. Of the remaining high-risk cases we found evidence for alternative telomere lengthening, thus bringing telomere maintenance into the focus of high-risk neuroblastomas. Five samples exhibited both, TERT rearrangements and MYCNamplifications. Initial and ongoing analyses have shown that these two events are subclonal in these samples, suggesting a parallel evolution of driver alterations.
Most recently, we extended our genomic analysis to 416 neuroblastoma cases using whole exome and targeted sequencing1. Together with the clinical data we were able to propose a new mechanism based risk classification of neuroblastoma (Figure 1). In this classification high-risk is exclusively characterized by the presence of telomere maintenance mechanisms (TERT rearrangements, MYCN amplifications, or ALT) otherwise the patient is at low risk to die from the disease. Furthermore, if RAS or p53 pathway mutations occurred on top of the telomere maintenance mechanisms patients did particularly poor. This group of patients therefore constitutes a very high-risk class. In total, we proposed a new classification of neuroblastoma based on molecular markers.
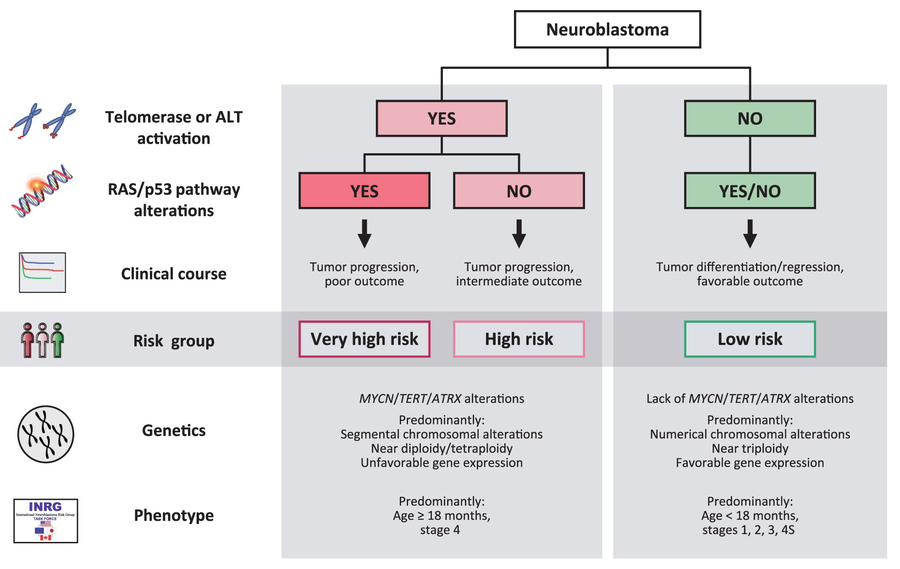
Figure 1: Proposed new classification of neuro-blastoma risk groups based on molecular markers (Ackermann et al., Science 2018).
We were able to include the modeling and analysis2of tumor evolution in the whole genome pan-cancer analysis of the International Cancer Genome Consortium (ICGC). This allows us to apply our concepts to about 2,800 cancer genomes across more than 30 different tumor types. The project has currently reached the publication phase.
By applying these/our developments to reconstruct the subclonal architecture we were able to compare the subclonal diversification between lung adenocarcinoma and small cell lung cancer. Here, the most limiting factor for this analysis was the relatively low sequencing coverage of 35X on average for the small cell lung cancer genomes. This hampers the detection of subclonal populations, especially in samples exhibiting low tumor purity or a higher ploidy. By systematically carrying a power analysis only about half of the 110 sequenced samples were qualified for this analysis. After establishing a concise measure to capture the mutational heterogeneity of the cancer genomes, we obtained that lung adenocarcinomas showed a 3-fold higher subclonal diversification5. This low degree of subclonal diversification is reflected in an almost perfect genomic match patient derived xenografts (derived from circulating tumor cells) with the primary tumor of the same patient.
By analyzing pairs of pre-treatment and relapsed chronic lymphocytic leukemia (CLL) patients that were treated with the BCL2 inhibitor venetoclax, we reconstructed the clonal dynamics towards resistance3. We observed convergent as well as divergent evolution in the 8 patients analyzed and found that BRAF and BTG1mutations are likely linked to the venetoclax resistance. Furthermore, alternative some mutations (BRAF and PDL1amplifications) point to further treatment options.
Recently we embarked into the reconstruction of tumor evolution from single cell sequencing data. This data allows to precisely reconstruct the phylogenetic relationship of the tumor samples. This can, e.g., be used to study genetic changes that lead to therapy failure due to drug resistance.
1. Ackermann S*, Cartolano M*,..., Peifer M*,Fischer M*. (2018) A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362, 1165-1170. *equal contribution
2. Cun Y, Yang TP, Achter V, Lang U, Peifer M. (2018) Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust.Nature Protocols 13, 1488-1501.
3. Herling CD, Abedpour N,..., Peifer M. (2018) Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nature Communications 9, 727.
4. Peifer M*,…, Fischer M*. (2015) Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700-4.*corresponding authors
5. George J, Lim JS,..., Peifer M*, Sage J*, Thomas RK*. (2015) Comprehensive genomic profiling of small cell lung cancer. Nature 524, 47-53. *corresponding authors

Dept. for Translational Genomics - CMMC Research Building
CMMC - PI - assoc. 16
show more…+49 221 478 96863
+49 221 478 97902
Dept. for Translational Genomics - CMMC Research Building
Robert-Koch-Str. 21
50931 Cologne
Nima Abedpour (PostDoc)
Maria Cartolano (PostDoc)
Milos Nikolic (PostDoc)
Stephani Pabel (PostDoc)
Sebastian Klein (PostDoc; Else-Kröner-Fresenius Program/from Pathology)
Lukas Maas (PostDoc; associated/from AG Thomas)
Armin Khonsari (PostDoc; associated/from AG Sos)
Tsun-Po Yang (doctoral student)
Lea Kowsky (doctoral student)
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC