Mitochondria are mobile organelles that constantly fuse and divide. These properties play a central role in quality control processes and energy expenditure. Mitochondrial fusion depends on the mitofusin proteins MFN1 and MFN2. Dysfunction of MFN2 causes the neuropathy Charcot-Marie-Tooth type 2A, is associated with Parkinson’s disease and with cardiovascular pathologies, but is also linked with type 2 diabetes and obesity and with non-alcoholic fatty liver disease and cancer. We aim at understanding at the molecular level the disease-underlying functions of MFN2.
Mitochondria are highly dynamic organelles which constantly adapt to metabolic cues via fusion and fission events. This plasticity is crucial for neuronal survival and is associated with aging-related diseases. The key effectors for mitochondrial fusion are the mitofusin proteins, MFN1 and MFN2. Deficiencies in MFN2 cause Charcot-Marie-Tooth disease (CMT), the most common degenerative disorder of the peripheral nervous system. Moreover, turnover of MFN2 by the proteasome is also linked to Parkinson’s disease, the most frequent movement disorder, and to cardiovascular diseases and diabetes.
We study at a molecular level the mechanisms allowing mitochondria to fuse or divide, via both positive and negative regulation of mitofusins. Under healthy conditions ubiquitin protects mitofusin from degradation and induces mitochondrial fusion, thus increasing mitochondrial interconnectivity. Instead, upon stress, additional ubiquitylation of mitofusins triggers their degra-dation by the proteasome, thus repressing mitochondrial fusion and leading to mitochondrial fragmentation by ongoing fission events.
In the presence of sufficient or excess nutrients, as in the case of stem and cancer cells but also in obesity and diabetes states, ATP and metabolite supply relies on glycolysis. This metabolic profile is associated to mitochondrial fragmentation, correlating with reduced levels of MFN2. In contrast, increased MFN2 levels inhibit tumor growth and reduce stemness, consistent with MFN2 being required for cellular differentiation.
However, mitochondrial dysfunction also plays a central role in non-mitotic cells heavily relying on oxidative phosphorylation (OXPHOS), like neurons. MFN2 mutations directly cause Charcot-Marie-Tooth type 2A (CMT2A), a peripheral neuropathy characterized by axonal degeneration and distal muscle atrophy. Several new CMT mutations identified so far show a widespread localization throughout the MFN2 protein (Fig. 1)
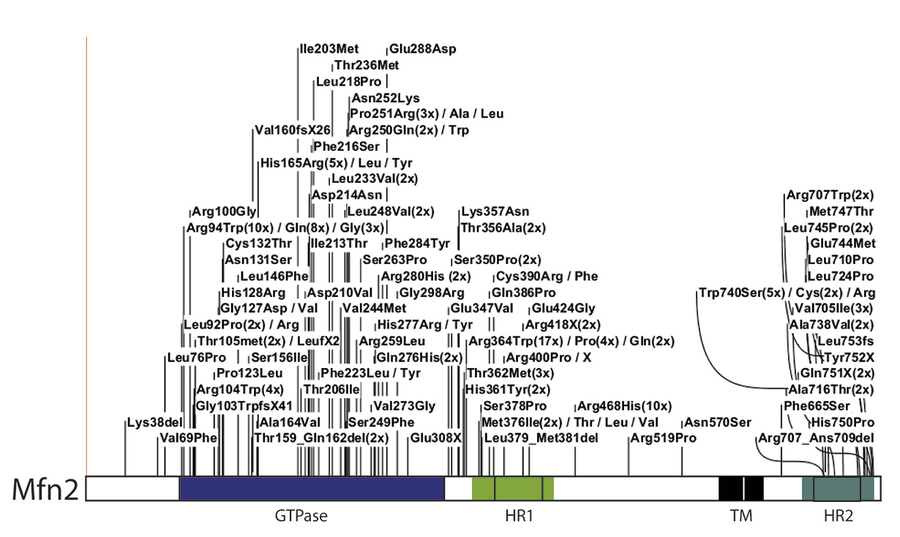
Figure 1.Mapping of the CMT2A-disease mutations identified in MFN2.
The mutations are depicted in the linear representation of MFN2, a dynamin-related GTPase protein, showing its additional domains: HR, heptad repeat; TM, transmembrane domain.
MFN2 is fundamental for efficient respiratory activity and ATP production, helping neurons to meet the high energy demands for proper function and preventing the distribution of dysfunctional mitochondria. Consistently, mitochondria are critical to the pathogenesis of the most common neurodegenerative diseases, like Alzheimer’s, Parkinson’s and Huntington’s , which have no cure.
Knock out of either one of the two mitofusins results in mitochondrial fragmentation (Fig. 2). In fact, the presence of fragmented mitochondria, correlating with decreased levels of MFN2 and presumably less fusogenic power, is a common and prominent early pathological feature of all these neuropathies. However, whether loss of fusion capacity per se or instead other functions of MFN2 are the critical disease-determinants is not known. Importantly, given that no effective therapies are available, it is particularly important to understand at the molecular level how MFN2 relates to these different pathophysiologic mechanisms.
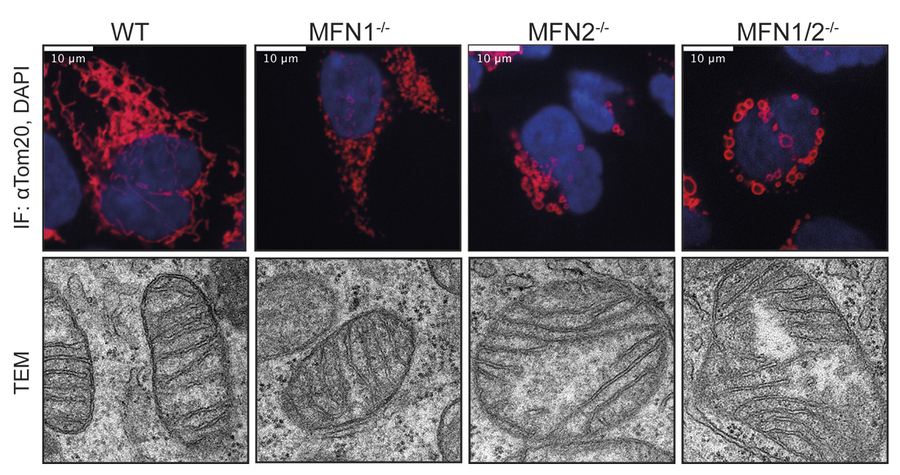
Figure 2. MFN2 alters mitochondrial morphology and ultrastructure. Immunofluorescence and transmission electron microscopy (TEM) of cells deficient for mitofusins.
Mitochondria are highly engaged in quality control and aging processes, mainly prevalent upon the accumulation over time of damaged organelles. The role of mitofusins in cellular stress responses, induced by excessive oxidation, has been described. It depends on the accumulation of ubiquitylated MFN1 and MFN2 proteins, among others, at the outer surface of mitochondria. This allows the recruitment of the E3 ubiquitin ligase Parkin to mitochondria and consequently signals the selective destruction of the damaged organelles.
Interestingly, human cells deleted for mitofusins presented several novel quality control-features. These involve both the ubiquitin-proteasome system and the lysosome, the two major pathways required for clearance of damaged proteins. Our findings ex-pand the role of MFN2 in signal-relay responses by ubiquitin-dependent chaperones or by the mito-phagy machinery.
Our long-term purpose is transferring from the basic molecular mechanisms to therapies of mitofusin-associated diseases. Importantly, divergence between our results and studies performed using mouse models highlight the importance of working with human cell models to study CMT2A and other pathologies linked to MFN2. Our results, using HEK293 and HeLa KO cell lines, will clearly allow starting to fill this gap.

Institute for Genetics
CMMC - PI - CAP 14
Mafalda.Escobar[at]uni-koeln.de
show more…+49 221 478 84257
+49 221 478 97295
Institute for Genetics
Institute for Genetics
50931 Cologne
http://www.uni-koeln.de/math-nat-fak/genetik/groups/Escobar/index.html
Mariana Joaquim
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC