The transcription factor nuclear factor erythroid 2-related factor 2 (NFE2L2/NRF2) is an established regulator of the cellular antioxidant response to xenobiotic and oxidative stress, thereby contributing to cytoprotection. However, similar to other cell survival pathways, NRF2 activation has a dark side, namely it protects malignant cells from death and promotes cancer cell growth, metabolic reprogramming, metastasis and resistance to cancer therapies. Under basal conditions, NRF2 is bound to Kelch-like ECH-associated protein 1 (KEAP1) that orchestrates its constant ubiquitination by E3 ligase Cullin 3 and its proteasomal degradation. Conditions that increase ROS production, such as the exposure to xenobiotics, radiation or metabolic stress, lead to oxidation of a number of cysteine residues on KEAP1 and disrupt the KEAP1-NRF2 interaction. As a result, NRF2 accumulates and moves to the nucleus, where it mediates transcription of its target genes (Fig. 1A). In addition, a non-canonical pathway of NRF2 activation, which depends on Sequestosome 1 (SQSTM1)/p62, has been described. Aberrant accumulation of p62, for instance upon impaired autophagy, and its phosphorylation on S351 strongly increases the affinity of p62 for KEAP1. This leads to KEAP1 sequestration and NRF2 release and nuclear translocation (Fig. 1A). However, p62 is a multifaceted protein with described functions in selective autophagy, cell death regulation, DNA damage response and NF-κB, mTORC1, ERK and K-RAS signaling pathways (Fig. 1B).
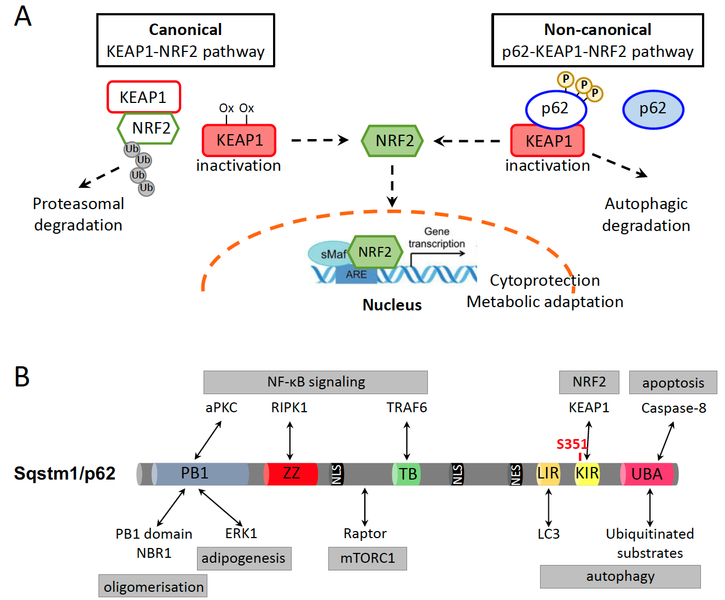
NRF2 activation is a well-documented oncogenic pathway. NRF2-activating and KEAP1-inactivating mutations have been identified in a substantial percentage of human cancers. Accordingly, we have recently shown that KEAP1 mutations were independent negative prognostic markers both for early- and late-stage NSCLC patients. On the other hand, p62 accumulation has also been reported in many cancer types and is linked with poor prognosis. This suggests that NRF2 activation through the non-canonical p62-KEAP1-NRF2 axis, which is independent of NRF2 or KEAP1 mutations, could be highly relevant in cancer progression and therapy resistance. Importantly, using a mouse model of hepatocarcinogenesis, we have recently shown that NRF2 activation alone was not enough to aggravate tumorigenesis. This indicates that deregulation of additional cellular events, including p62 accumulation and autophagy, cooperate with the NRF2 pathway and are required to unleash its oncogenic potential.
In the era of personalized oncology and with the number of targeted treatments growing, therapeutic decisions depend on detailed molecular subtyping. Our research aims at assessing the prevalence of p62-KEAP1-NRF2 axis activation across various cancer types and whether its activation correlates with other cellular and signaling pathway markers. To find potential therapeutic targets for tumors characterized by activation of this pathway, our goal is also to identify kinase inhibitors that impair p62 phosphorylation and lead to decreased NRF2 activation.
In this project, we will address our aims with the following approaches:
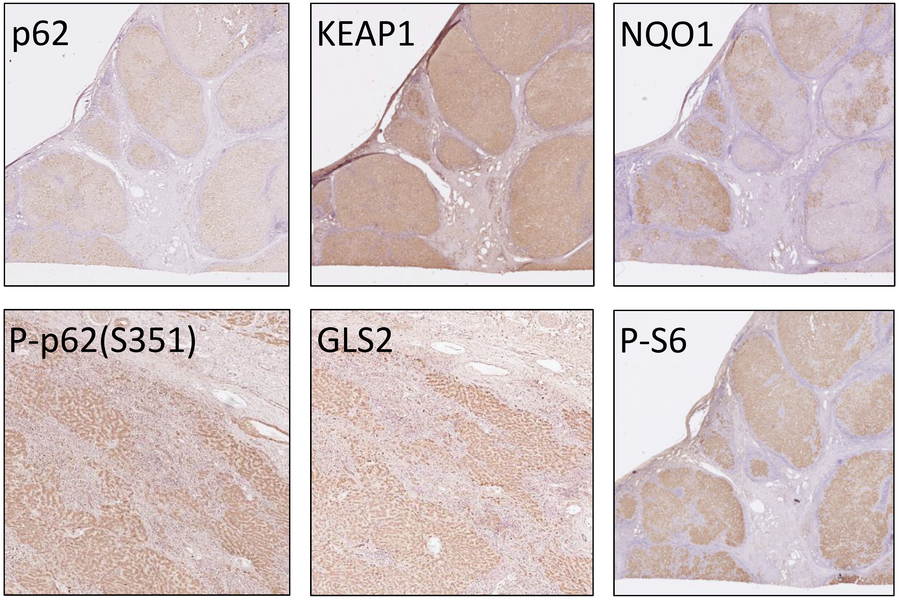
Mice with hepatic impairment of autophagy have been shown to develop chronic liver injury and liver tumors in an NRF2- and p62-dependent manner. Accordingly, we have generated ATG16L1LPC-KO mice that show autophagy inhibition in hepatocytes, as well as strong p62 accumulation and phosphorylation at S351. These mice exhibit spontaneous liver damage, inflammation, fibrosis, hepatomegaly and eventually develop liver cancer (Figure 3A-C) (Kondylis et al., 2022). As anticipated, additional p62 deficiency (p62-/-) significantly prevented liver injury and ameliorated hepatomegaly and hepatocarcinogenesis (Figure 3B-C).
In search for potentially relevant biomarkers to be included in our IHC screening panel, we performed a quantitative proteomics analysis in liver lysates from WT, p62-/-, ATG16L1LPC-KO and ATG16L1LPC-KO p62-/- mice. We focused on proteins that are upregulated in ATG16L1LPC-KO livers and whose expression either became normalized in the absence of p62 and NRF2 activation or remained upregulated in ATG16L1LPC-KO p62-/- mice (Figure 3D). In line with the previously reported correlation between the NRF2 pathway and ferroptosis, we found high expression of several ferroptosis antagonists in ATG16L1-deficient livers, suggesting that ferroptosis inducers in p62-KEAP1-NRF2-positive tumors may elicit an anti-tumor effect. Our data also showed evidence of increased glutaminolysis, a pathway that is also linked to NRF2 activation and turns glutamine into TCA metabolites to promote cancer cell metabolic adaptation. The effect of targeting these two pathways in hepatocarcinogenesis will be tested in ATG16L1LPC-KO mice.

Development of biomarker combinations is necessary to define molecular subtypes in cancer, as this will facilitate therapeutic decisions and may improve their outcome. Our project intends to develop such an IHC profile for tumors that are characterized by NRF2 activation induced by p62 accumulation, which may result from defects in autophagy and can affect additional oncogenic signaling pathways. Besides systematically mapping the penetrance of this pathway in 5 common cancer types, we aim to identify clinically-relevant kinase inhibitors that prevent p62 phosphorylation, dampen NRF2 activation and are likely to render cancer of this molecular subtype more susceptible to cytotoxic anti-cancer treatments.

Institute for General Pathology and Pathological Anatomy
CMMC - PI - A 03
CMMC - Co-PI - A 06
Executive Board Member
reinhard.buettner[at]uk-koeln.de
show more…+49 221 478 6320
+49 221 478 6360
Institute for General Pathology and Pathological Anatomy
Kerpener Str. 62
50937 Cologne

Institute of Pathology
CMMC - Co-PI - A 03
show more…+49 221 478 84362
Institute of Pathology
https://pathologie.uk-koeln.de/forschung/nachwuchsgruppen/molekulare-hepatologie-ag-kondylis/
Scientific Coordinator
Dr. Sonja Meemboor
PostDoc
Dr. Ka-Won Noh
Medical doctor
Tillmann Bedau
PhD student
Marta Pistone
Technician
Hannah Eischeid-Scholz
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC