Hereditary spastic paraplegia (HSP) is an inherited progressive neurological condition characterized by weakness and spasticity of the lower limbs, owing to the selective degeneration of axons of corticospinal motoneurons (CSMNs). No therapy, besides symptomatic treatment, is currently available for HSP patients, who experience progressive difficulties in ambulation, ultimately leading to wheelchair confinement. Our group has a long-standing interest in defining the pathogenic pathways implicated in HSP, and has developed several mouse models for the disease. A big challenge in the field is to pinpoint pathogenic commonalities among HSP forms of different genetic origin, in order to identify shared therapeutic approaches for HSP patients. In this project we investigate whether degeneration of CSMN axons in HSP is caused by unifying neuronal dysfunctional pathways or local activation of axonal death cascades. To this end, we will perform high-throughput unbiased analyses across four representative forms of HSP (SPG4, SPG7, SPG18, and SPG62), using integrated transcriptomics, proteomics, lipidomics and metabolomics in established mouse models. Analyses will be performed ex vivo on the spinal cord and other relevant tissues and in vitro on neuronal soma and axons isolated by means of microfluidic chambers. Identified pathways will be further explored in cellular models and probed for cross-species conservation on HSP patients’ biomaterial. Our ultimate goal is to identify and test convergent druggable targets to prevent or block disease progression in genetically different forms of HSP.
Advances in understanding the genetic bases of HSP have highlighted the complexity of the disease pathogenesis and failed to reveal a common pathogenic mechanism. This hampers attempts to find a unifying therapeutic approach for seemingly different disease forms. Our study will allow:
1) to identify shared early-onset pathogenic pathways among different forms of HSP;
2) to discover if the mechanism of axonal death is conserved;
3) to instruct drug-repurposing strategies as novel therapeutic approaches.
In the shorter term, we will translate our results to preclinical trails in the mouse. The long-term perspective is to validate any disease biomarker and pathogenic pathway identified in this study in HSP patient-derived cells and body fluids.
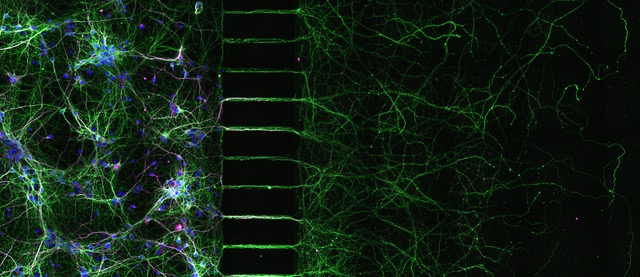
To identify HSP-specific signaling, stress, and death pathways in the neuronal population of interest, the CSMNs and their axons, we employ both ex vivo and in vitro approaches. We make use of genetic mouse models for different forms of HSP that exemplify distinct pathogenic pathways (mitochondrial dysfunction in SPG7; cytoskeletal and lipid droplet impairment in SPG4; disturbances of endoplasmic reticulum-associated degradation and Ca2+ signaling in SPG18 and SPG62). We will explore transcriptional, proteome and metabolome changes in the motor cortex and the spinal cord during the progression of the disease. In addition, we have established compartmentalized axonal cultures to exquisitely separate axons from the neuronal soma, and identify potential axono-centric mechanisms of degeneration. Finally, we aim to confirm identified pathways and processes in biological material from human HSP patients and test molecular mechanisms in cellular model systems.
Montoro-Gámez, C., Nolte, H., Molinié, T., Evangelista, G., Tröder, S., Barth, E., Popovic, M., Trifunovic, A., Zevnik, B., Langer, T., Rugarli E.I. (2023). SARM1 deletion delays cerebellar but not spinal cord degeneration in an enhanced mouse model of SPG7 deficiency. Brain. 2023 Apr 22:awad136. doi: 10.1093/brain/awad136. Online ahead of print. PMID: 37086482
Tadepalle, N. and Rugarli, E.I. (2021). Lipid Droplets in the Pathogenesis of Hereditary Spastic Paraplegia. Front. Mol. Biosci., 10 May 2021. 8:673977. doi: 10.3389/fmolb.2021.673977. eCollection 2021. Review
Tadepalle, N., Robers, L. Veronese, M. Zentis, P. , Babatz, F. Brodesser, S. Gruszczyk, A.V., Schauss, A., Hoening, S., and Rugarli, E. I. (2020). Microtubule-dependent and independent roles of spastin in lipid droplet dispersion and biogenesisLife Sci. Alliance 3 (6):e202000715.
Murru, S., Hess, S., Barth, E., Almajan, E.R., Schatton, D., Hermans, S., Brodesser, S., Langer, T., Kloppenburg, P. and Rugarli, E.I. (2019). Astrocyte‐specific deletion of the mitochondrial m‐AAA protease reveals glial contribution to neurodegeneration. Glia 67(8):1526-1541.
Wang, S., Jacquemyn, J., Murru, S., Martinelli, P., Barth, E., Langer, T., Niessen, C.M., and Rugarli, E.I. (2016). The Mitochondrial m-AAA Protease Prevents Demyelination and Hair Greying. PLoS Genet 12(12): e1006463.
Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jungst C, Tzschichholz A, Schauss A, Honing S, Trifunovic A, Daga A, and Rugarli E.I. (2015). Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet 11, e1005149.
Kondadi, A.K., Wang, S., Montagner, S., Kladt, N., Korwitz, A., Martinelli, P., Herholz, D., Baker, M.J., Schauss, A.C., Langer, T. and Rugarli, E.I. (2014). Loss of the m-AAA protease subunit AFG3L2 causes mitochondrial transport defects and tau hyperphosphorylation. EMBO J, 33, 1011-1026.

CECAD Cologne & Institute for Genetics
CMMC - PI - C 14
show more…+49 221 478 84244
+49 221 478 97902
CECAD Cologne & Institute for Genetics
Joseph-Stelzmann-Str. 26
50931 Cologne
PostDocs
Alexander Kaczmarek, Thibaut Molinié, Marta Zaninello
PhD students
Irene Costa, Giada Di Pietro,
Giovanna Evangelista, Rrejusha Parayil,
Tim Schlegel, Matteo Veronese
Master students
Alessia Lofrano
Technician
Esther Barth
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC