We are interested in understanding the mechanisms governing the homeostasis of the cellular proteome (proteostasis). Protein structures are highly complex and dynamic, and several stresses can compromise their integrity. Therefore, cells are equipped with an intricate and adaptive network of factors ensuring protein synthesis, folding, trafficking, conformational maintenance, and degradation. Importantly, misfolded proteins are not only dysfunctional, but they can also coalesce into potentially toxic aggregates. These represent a hallmark of many aging-associated neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases. Our aims are to understand how protein quality control pathways distinguish defective from functional proteins, how the decision between repair and elimination is made, and how these processes are activated under stress and pathological conditions.
Proteins represent the functional units of the cell. To perform a task, they must adopt specific three-dimensional structures, or folds. Folding requires proteins to navigate a complex energy landscape, putting them at risk of adopting non-native structures. Moreover, a myriad of cellular stresses ensued by environmental, metabolic, and pathogenic conditions can promote unfolding and aggregation of metastable proteins. Correct folding is ensured by different classes of chaperone proteins that guide clients along productive folding pathways. When such system fails, terminally misfolded proteins must be rapidly eliminated in order to prevent formation of toxic aggregates, a task that is carried out by a large number of factors belonging to the ubiquitin-proteasome and autophagy–lysosome systems. Importantly, protein quality control effectors are also involved in further aspects of proteome balance, such as regulation of protein abundance, subcellular localization, and assembly into macromolecular complexes. Our research group aims at understanding how protein quality control pathways can shape cell physiology, and how its factors can achieve client specificity given the enormous heterogenicity of the proteome.
Production of functional proteins also requires the correct flow of information from DNA into RNA and finally into protein. The underlying processes – DNA duplication, transcription, RNA maturation, and translation all involve intrinsic levels of error, and therefore are under scrutiny of proof-reading quality control pathways. In recent years it became evident that translation plays a fundamental role in the detection of defective mRNAs. For example, translation of mRNA species lacking stop codons results in ribosomal stalling, which serves as signal for the recruitment of both the non-stop mRNA decay (NSD) and the ribosome-associated protein quality control (RQC) pathways. Translation of defective non-stop mRNAs results in truncated proteins that are likely to be dysfunctional. The RQC pathway ensures that those aberrant species are committed to degradation, avoiding possible dominant negative effects of proteins lacking functional domains, and avoiding unnecessary burden to the post-ribosomal protein folding machinery. We have described that the RQC quality control pathway is unique in that degradation is largely independent of the folding state of its client proteins, which has important implications for the role of RQC in the production of antigenic peptides (Figure 1).
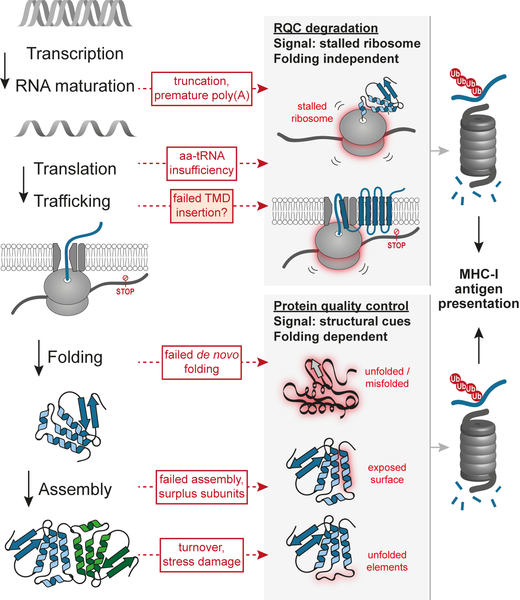
A mouse model harboring a hypomorphic mutation in LTN1, encoding for the Listerin E3 ligase responsible for the ubiquitination of the stalled nascent chains, exhibits profound early-onset and progressive neurological and motor dysfunction. In yeast, failure of the RQC machinery can result in protein aggregation and disruption of the proteostasis network. Major progress has been made towards understanding the mechanistic features of RQC components, and why its clients tend to aggregate upon RQC failure. Although studies using synthetic constructs have described a variety of potential triggers for ribosomal stalling, the endogenous sources of elongation stalls underlying the neurodegenerative phenotype remain to be fully characterized. Our future goals include understanding how stalling-inducing defective mRNAs are produced, and how stress conditions can influence their formation.
Using MHC-I antigen presentation as a proxy for proteasomal degradation, we have obtained a dataset of endogenous substrates of RQC in human cells grown under physiological conditions. The analysis confirmed the previous notion that erroneous cleavage and polyadenylation of mRNAs within the coding sequence is an important trigger of RQC in vivo. Surprisingly, however, the analysis also showed that proteins containing large numbers of transmembrane domains (TMDs) have higher tendencies to be degraded by RQC. Therefore, in the future we will investigate the relationship between ribosomal elongation and co-translational insertion of proteins into membranes.
For further information please check the Trentini Lab - Protein quality control and stress response webpage.

Center for Molecular Medicine Cologne | Lab. of Protein Quality Control and Stress Response | CMMC Research Building
CMMC - PI - assoc. JRG 05
show more…+49 221 478 42566
Center for Molecular Medicine Cologne | Lab. of Protein Quality Control and Stress Response | CMMC Research Building
Robert-Koch-Str. 21
50931 Cologne
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC