The development of massively parallel sequencing has fueled genome-centric cancer research. This technology has enabled us to sequence entire cancer genomes and transcriptomes in a relatively short period of time. However, the data produced by this approach is highly complex and difficult to analyze in order to extract the biologically relevant information. Within this project we systematically integrated genome with transcriptome data to identify these genomic alterations.
The sequencing of entire cancer genomes provides a comprehensive portrait of the genomic alterations that have occurred during tumorigenesis. The most common changes are subtle sequence alterations (simple substitutions, small insertions and deletions) followed by structural changes such as copy number alterations and genomic rearrangements. On a genome-wide scale, the number of all these alterations is typically within the range of 104–106 events and varies significantly between tumor types with pediatric tumors at lower end of the spectrum and lung cancers, melanoma are being the most mutated cancer genomes. However, most of these mutations are likely to have no functional relevance to the cancer cells. Thus, only a minority of mutations is biologically relevant and can be associated to the pathogenesis of the cancer cells. Finding these genome alterations from sequencing data of large collectives of patient samples is still a challenging task.
Over the past years we have developed a cancer genome analysis toolkit to detect all major genome alterations (point mutations, copy number changes, and rearrangements) and applied it to several large-scale sequencing projects1-4. Furthermore, we developed a series of downstream analysis methods to identify relevant genome alterations across a series of sequenced tumors1-5.
Within this project, we systematically integrated genome with transcriptome data to further identify relevant genome alterations. We applied our approach to large-scale sequencing efforts of small cell lung cancer and neuroblastoma.
About 16% of all lung tumors accounts for small cell lung cancer. Clinically, most of the patients respond well on conventional chemotherapy but relapse quickly with a chemo-resistant phenotype leading to a 5-year survival of less than 5% of the patients. Genomically, small cell lung cancer is characterized by a bi-allelic loss of the two tumor suppressors: TP53 and RB12.
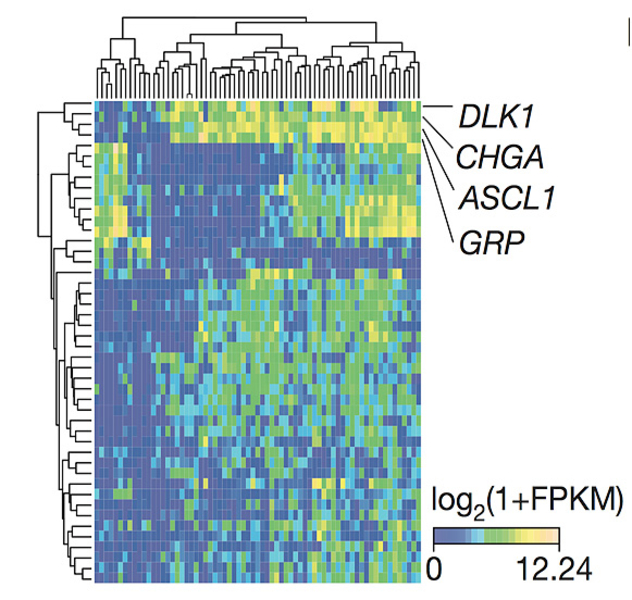
To draw a comprehensive molecular portrait of primary small cell lung cancer tumors, we sequenced whole genomes of 110 patient samples along with the transcriptomes of 69 specimens. By inferring robust gene expression changes within this collective of tumors, we found that differentially expressed genes are linked to the neuroendocrine differentiation of small cell lung cancer (CHGA, GRP) and to the Notch signaling pathway (DLK1, ASCL1; Figure 1).
In addition, we found an accumulation of damaging mutations in NOTCH-family genes, thus suggesting together with the gene expression analysis a tumor suppressive role of NOTCH in the context of small cell lung cancer. Overexpressing NOTCH2 in a Trp53, Rb1, Rbl1 conditional triple-knockout small cell lung cancer mouse model showed a significantly prolonged tumor development and fewer tumors, when compared to triple knockout mice without NOTCH2 overexpression2. This, therefore, validates the tumor suppressive role of NOTCH in small cell lung cancer. Furthermore, inhibition of the NOTCH negative regulator DDL3 showed massive therapeutic responses in small cell lung cancer patients (Pietanza et al., ESMO 2015).
Neuroblastoma is the most common pediatric solid tumor arising from cells of the neural crest. Clinically, neuroblastoma shows a diverse outcome that ranges from spontaneous regression to an unfavorable clinical course. The most common genomic alteration is the amplification MYCN, which is associated with a high risk to die from the disease.
We sequenced the whole genomes of 56 primary neuroblastomas with the aim to find novel relevant genome alterations. After applying our method to detect recurrent genomic rearrangements, we found an enrichment of translocations in the region of the telomerase reverse transcriptase gene TERT1. These rearrangements were only present in high-risk cases and were mutually exclusive with MYCN amplifications, suggesting a possible convergence on similar effector functions.
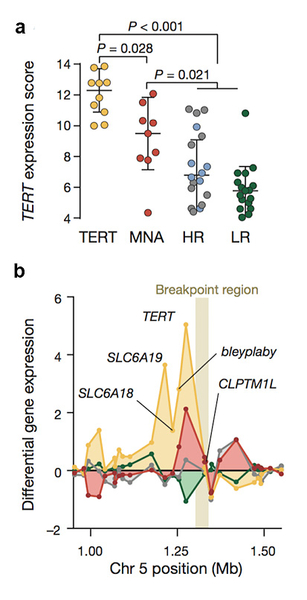
By analyzing the gene expression of TERT between cases harboring these rearrangements, MYCN amplifications, and the remaining high- and low risk cases, we found that only TERT rearrangements and MYCN amplifications lead to a transcriptional up-regulation of TERT (Figure 2a). Furthermore, the differential analysis of the surrounding genes revealed that other genes in the TERT locus are also up regulated in the presence of the rearrangement, whereas only TERT was up regulated in the samples bearing MYCN amplifications (Figure 2b).
This suggests that the TERT rearrangements are bringing regulatory elements in the region of TERT leading to the observed transcriptional up-regulation. In fact, ChIP-seq of the histone mark H3K27ac confirmed that strong transcriptional enhancers were translocated to the region1. Finally, we found that the remaining high-risk cases lacking both MYCN amplifications and TERT rearrangements showed evidence of the presence of alternative telomere lengthening mechanisms. Therefore, we conclude that telomere maintenance mechanisms are a hallmark of high-risk neuroblastoma.
We currently embarked into the reconstruction of tumor evolution from sequencing data. The developed methods are currently applied to series of chronic lymphocytic leukemia samples and are part of the Pan-Cancer Analysis of the International Cancer Genome Consortium. Here, about 2,800 whole cancer genomes across more than 30 tumor types are currently being analyzed.
1 Peifer M*, Hertwig F,..., Fischer M*. (2015) Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700-704. *corresponding authors
2 George J,..., Peifer M*, Sage J*, Thomas R*. (2015) Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47-53. *corresponding authors
3 Peifer M, Fernandez-Cuesta L, Sos M, et al. (2012). Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature Genetics 44, 1104-1110.
4 Fernandez-Cuesta L*, Peifer M*, Lu X et al. (2014). Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nature Communications 5, 1-7. *equal contribution
5 Lu X, Thomas R, and Peifer M (2014). CGARS: Cancer Genome Analysis by Rank Sums. Bioinfomatics 30, 1295-1296.
6 Fernandez-Cuesta L, Sun R, Menon R, et al. (2015) Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biology 16, 7.
Information from this funding period will not be updated anymore. New research related information is available here.

Dept. for Translational Genomics - CMMC Research Building
CMMC - PI - assoc. 16
show more…+49 221 478 96863
+49 221 478 97902
Dept. for Translational Genomics - CMMC Research Building
Robert-Koch-Str. 21
50931 Cologne
Yupeng Cun (PostDoc)
Maria Cartolano (PostDoc)
Nima Abedpour (PostDoc)
Tsun-Po Yang (doctoral student)
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC