The functional integrity of mitochondrial depends on a defined lipid composition of its membranes. Disturbances in the metabolism of cardiolipin, a constituent of mitochondrial membranes, are associated with various cardiomyopathies including Barth syndrome, Sengers syndrome and dilated cardiomyopathy with ataxia (DCMA). We are analysing mitochondrial membrane scaffold proteins, intramitochondrial phospholipid trafficking and metabolism to unravel the pathogenic mechanisms underlying these diseases.
Mitochondrial membranes have a characteristic lipid composition, which is required for its function. The diphosphatidyl glycerol phospholipid cardiolipin (CL) has been recognized as a ubiquitous signature lipid of mitochondria where it preserves mitochondrial structure and function. CL oxidation and an impaired CL accumulation have been observed during aging and cause various diseases. The first genetic disorder that was associated with an impaired CL metabolism is Barth syndrome (BTHS), an X-linked recessive cardiomyopathy. Barth syndrome is characterized in affected males by cardiomyopathy, neutropenia, skeletal myopathy, and prepubertal growth delay. Disease-causing mutations lead to the loss of the transacylase tafazzin in the mitochondrial intermembrane space, which is involved in the remodeling of CL acyl chains. Loss of tafazzin results in decreased accumulation of CL in the mitochondrial membranes, impairs OXPHOS function, and disturbs the ultrastructure of mitochondria. Similarly, the function of the mitochondrial acyl glycerol kinase AGK has been linked to the mitochondrial CL metabolism. Mutations in AGK cause cardiomyopathy and are associated with Sengers syndrome, an autosomal recessive disease characterized by congenital cataract, hypertrophic cardiomyopathy, skeletal myopathy and lactic acidosis. Moreover, mutations in the mitochondrial co-chaperone DNAJC19, a DnaJ-like chaperone protein and predicted component of the mitochondrial presequence translocase, cause dilated cardiomyopathy with ataxia (DCMA), an early onset dilated cardiomyopathy syndrome with clinical symptoms similar to BTHS. These genetic data suggest similarities in the pathogenesis of these cardiomyopathies.
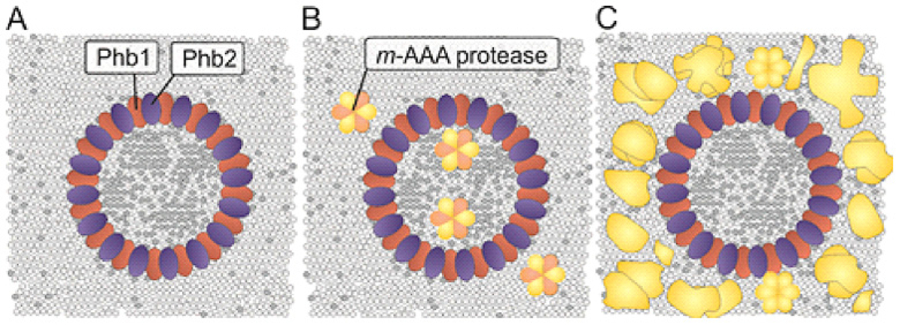
Our group examines mechanisms contributing to the phospholipid homeostasis in mitochondrial membranes, focusing on lipid transport processes between both membranes and the function of scaffold proteins, such as prohibitins, as membrane organizing complexes in the inner membrane. Prohibitins form ring complexes with a diameter of ~20 nm, which are thought to act as scaffolds for both proteins and phospholipids (Figure 1). Prohibitins are essential for mitochondrial function and cell survival. Genetic evidence in yeast links their function to mitochondrial phospholipids such as phosphatidyl ethanolamine and cardiolipin. We have shown that tissue-specific ablation of prohibitins in the mouse lead to neuro-degeneration, diabetes and kidney failure.
We have determined the interactome of prohibitins in mitochondria using affinity purification of combined with quantitative mass spectrometry and identified DNAJC19 mutated in DCMA as the strongest binding partner of prohibitins.
The similarities in the clinical presentations of DCMA and Barth syndrome patients and the putative function of prohibitin complexes as scaffolds for phospholipids prompted us to examine a role of DNAJC19 in the mitochondrial CL metabolism. Combining loss-of-function studies with quantitative lipidomics, we observed an altered spectrum of CL species in mitochondrial membranes lacking DNAJC19, reminiscent of BTHS patients lacking tafazzin. These results suggest that disturbed CL remodeling is of pathogenic relevance in both, BTHS and DCMA. How prohbitin-DNAJC19 complexes affect CL acylation is presently not understood, but it is an attractive possibility that they modulate directly CL remodeling by tafazzin (Figure 2).
Sengers syndrome is caused by mutations in the acyl glycerol kinase AGK, a mitochondrial kinase phosphorylating monoacylglycerol and diacylglycerol to lysophosphatidic (LPA) and phosphatidic acid, respectively. Accordingly, AGK was proposed to regulate cell signalling via LPA and to allow synthesis of CL along an alternative route. Accumulation of the ATP/ADP carrier in the inner membrane was observed in patient cells, but the molecular basis of this observation remained obscure. In order to elucidate pathogenic mechanisms in Sengers syndrome, we began to examine how disease-associated mutations affect AGK function and analyse the function of AGK for the mitochondrial CL metabolism.
We will continue our studies to unravel the pathogenic mechanisms underlying three related but distinct cardiomyopathies, for which there is currently no therapy available. Focusing on the metabolism of CL, our studies will provide general insight into the role of this mitochondrial signature lipid for mitochondrial function.
Aaltonen, M.J., Friedman, J.R., Osman, C., Salin, B., di Rago, J.-P., Nunnari, J., Langer, T., and Tatsuta, T. (2016) MICOS and phospholipid transfer by Ups2-Mdm35 organize membrane lipid synthesis in mitochondria. J. Cell Biol. 213, 525-534.
Richter-Dennerlein, R., Korwitz, A., Haag, M., Tatsuta, T., Dargazanli, S., Baker, M.J., Decker, T., Lamkemeyer, T., Rugarli, E.I., and Langer, T. (2014) DNAJC19 associated with cardiomyopathy assembles with prohibitins in a complex regulating cardiolipin remodeling. Cell Metab. 20, 158-171.
Potting, C., Tatsuta, T., König, T., Haag, M., Wai, T., Aaltonen, M.J., and Langer, T. (2013) TRIAP/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 18, 287-295.
Connerth, M., Tatsuta, T., Haag, M., Klecker, T., Westermann, B., and Langer, T. (2012) Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815-818.
Potting, C., Wilmes, C., Engmann, T., Osman, C., and Langer, T. (2011) Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 29, 2888-2898.
Osman, C., Haag, M., Wieland, F.T., Brügger, B., and Langer, T. (2010) A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 29, 1976-1987.
Osman, C., Haag, M., Potting, C., Rodenfels, J., Dip, P. V., Wieland, F. T., Brügger, B., Westermann, B., and Langer, T. (2009) The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regula¬tors in mitochondria. J. Cell Biol. 184, 583-596.
Information from this funding period will not be updated anymore. New research related information is available here.
Milena Vukotic (PostDoc)
Takashi Tatsuta (PostDoc)

Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC