Spinal muscular atrophy (SMA) is caused by homozygous absence of the survival motor neuron gene 1 (SMN1) and inability of SMN2 to compensate for the SMN1 loss. We identified plastin 3 (PLS3) as a protective genetic modifier of SMA, which is able to counteract SMA development in humans. Using various animal models (fish and mouse) we demonstrate that PLS3 overexpression efficiently protects SMA across species. Moreover, the genetic modifier PLS3 allowed us to unravel endocytosis as one of the main impaired cellular pathomechanisms in SMA, which is fully rescued in vitro and in vivo by PLS3 overexpression. Moreover, we demonstrate that a combinatorial therapy based on PLS3 overexpression and low dose SMN antisense oligonucleotides (ASOs) can rescue the severe SMA phenotype in mice, opening a new therapeutic perspective for SMA.
Autosomal recessive proximal SMA, one of the most common and devastating motor neuron disorders in humans is caused by a deficiency in SMN, a protein involved in a plethora of different cellular mechanisms including splicing, translation regulation, axonal transport, stress response etc. The exact underlying cellular pathway, which mainly disturbs neuromuscular synapses is still unknown. Based on expression studies in rare families with phenotypically discordant siblings - affected and asymptomatic - we identified plastin 3 (PLS3), a cytoskeletal protein involved in F-actin dynamics, to be highly upregulated in asymptomatic but not symptomatic SMN1-deleted siblings: PLS3 was therefore considered as an excellent candidate SMA protective modifier (6). Functional studies in various cellular systems and in zebrafish demonstrated that PLS3 overexpression rescues the axonal and NMJ defects caused by reduced SMN levels (5, 6).
The main findings of this funding period are:
We found high PLS3 expression in lymphoblastoid cell lines but not in fibroblasts of asymptomatic patients as compared to symptomatic, SMA III-affected siblings. To find out whether PLS3 is differentially expressed in motor neurons of asymptomatic individuals, we generated induced pluripotent stem cells (iPSCs) from fibroblasts of two discordant SMA families including three asymptomatic and three SMA type III patients and compared these to iPSCs from SMA type I and control individuals. After validation of pluripotency in iPSC lines, motor neurons were differentiated from iPSC-derived small molecule neural precursor cells (smNPCs). PLS3 was highly upregulated only in motor neuron cultures of asymptomatic but not symptomatic patients suggesting a tissue-specific regulation of PLS3. Visible accumulation of PLS3 in the shaft and rim of growth cones in motor neuron cultures from asymptomatic patients implied an important role of this protein in neuromuscular synapse formation (1).
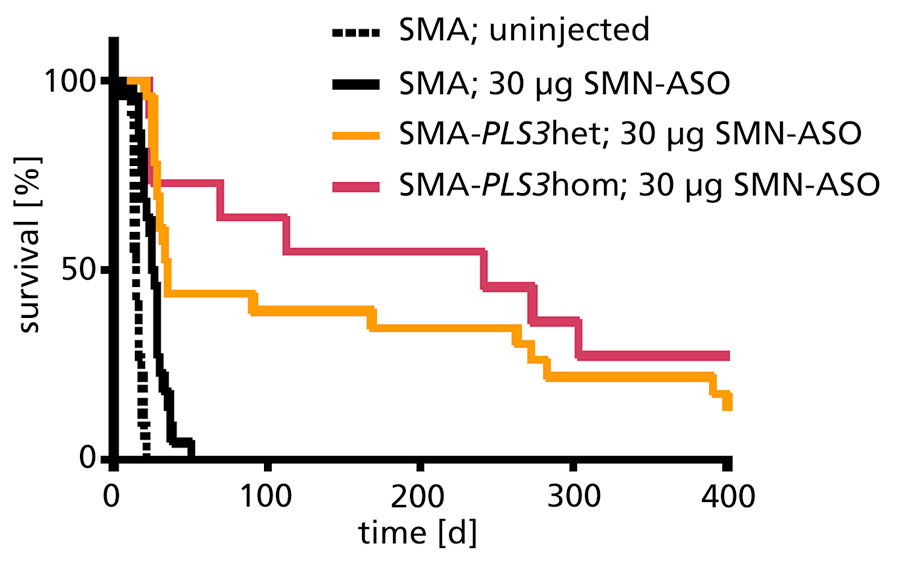
Previously we have generated a transgenic mouse model overexpressing PLS3 within the ROSA26 locus under a chicken beta-actin promoter. In a severe SMA model, PLS3 overexpression rescued the motor abilities by restoring F-actin dependent processes at the neuromuscular junction level but had minor impact on survival of the mice (5). This is due to impairment of most inner organs in this severe SMA animal model, showing that PLS3 alone cannot overcome the severe lack of SMN protein. Now, we established an intermediate SMA mouse model by injecting a suboptimal dose of SMN-ASOs that corrects SMN2 splicing. SMN-ASOs are currently successfully used in several clinical trials (IONIS/Biogen). We demonstrate that the combinatorial effect of suboptimal SMN-ASO treatment and PLS3 overexpression – a situation resembling the human condition in asymptomatic SMN1-deleted individuals - rescues survival (from 14 to >250 days; Fig. 1) and motoric abilities of a severe SMA mouse model (2). These data emphasize the power of combinatorial therapy based on SMN2 splice correction and PLS3 overexpression to efficiently treat SMA.
We identify impaired endocytosis as a major cellular mechanism underlying SMA, which is restored to normal levels by PLS3 overexpression. Upon low frequency electro-stimulation, endocytic FM1-43 uptake in the presynaptic terminal of neuromuscular junctions is restored to control levels in SMA-PLS3 mice. Moreover, proteomics and biochemical analysis revealed coronin 1C (CORO1C), another F-actin binding protein, which directly binds PLS3 in a Ca2+-dependently manner. Similar to PLS3, CORO1C overexpression restored fluid-phase endocytosis in SMN knockdown cells by elevating F-actin levels and rescued the axonal truncation and branching phenotype in Smn-depleted zebrafish (2). Our findings emphasize the power of genetic modifiers to unravel the cellular pathomechanisms underlying SMN deficiency in SMA.
To further decipher the molecular pathway how PLS3 overexpression rescues the SMA phenotype, we searched for PLS3 interacting partners using tandem tag purification followed by mass spectrometry. HEK293T cell lines stably overexpressing Flag/His-tagged PLS3 or the vector alone were generated and Co-IP eluates analysed by mass spectrometry. 104 proteins were identified as potential PLS3 binding partners. Among different protein categories which were identified, two F-actin binding proteins (CORO1C and TMOD3) were considered of particular interest and analysed in further detail. We confirmed the interaction between PLS3 and both proteins by Co-IP. Pull-down assays with full-length or deletion constructs unravelled the exact region of interaction between PLS3 and CORO1C. Most interestingly, this interaction was calcium-dependent.
The actin cytoskeleton has been shown to have an important role in axon growth and pathfinding as well as in the transport and recycling of vesicles in the presynapse. Immunocytochemical stainings showed that PLS3 co-localizes with both proteins, although in different structures. Staining of primary motor neurons isolated from wild-type and SMA mice showed the co-localization of PLS3 and PLS3-CORO1C in both axons and growth cones. To functionally characterize the role of both proteins in the axon pathology of SMA, CORO1C or TMOD3 mRNA were microinjected with smn morpholino in zebrafish. Interestingly, only CORO1C but not TMOD3 rescued the axonal outgrowth (2).
Meanwhile, we identified a second SMA modifier, which together with PLS3 strengthen endocytosis as the major cellular pathomechanism of SMA. Based on these new exciting findings we seek for novel therapeutic possibilities and deeper understand the endocytic defects in SMA.
*equally contributed first authors
Information from this funding period will not be updated anymore. New research related information is available here.

Institute of Human Genetics - CMMC Research Building
CMMC - PI - C 18
CMMC - Executive Board Member
brunhilde.wirth[at]uk-koeln.de
show more…+49 221 478 86464
+49 221 478 86465
Institute of Human Genetics - CMMC Research Building
Kerpener Str. 34
50931 Cologne
https://humangenetik.uk-koeln.de/forschung/ag-neuromuskulaere-und-skelettale-erkrankungen/
Seyyedmohsen Hosseinibarkooie (Postdoc)
Markus Riessland (PostDoc, currently at Rockefeller Univ.)
Markus Storbeck (PostDoc)
Laura Torres-Benito (PostDoc)
Ludwig Heesen (PhD student, completed)
Christian Hoffmann (MD student)
Eva Janzen (PhD student)
Anna Kaczmarek (PhD student, completed)
Mert Karakaya (MD student)
Lilian Martinez (PhD student)
Natalia Mendoza-Ferreira (PhD student)
Janine Milbradt (PhD student)
Miriam Peters (PhD student)
Svenja Schneider (PhD student)
Eike Strathmann (PhD student)
Aaradhita Upadhyay (PhD student)
Andrea delle Vedove (MD/PhD student)
Vanessa Grysko (technician)
Irmgard Hölker (technician)
Kristina Hupperich (technician)
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC