High-fat diet (HFD) feeding induces rapid reprogramming of systemic metabolism. Here, we demonstrate that HFD feeding of mice downregulates glucose transporter (GLUT)-1 expression in blood-brain barrier (BBB) vascular endothelial cells (BECs) and reduces brain glucose uptake. Upon prolonged HFD feeding, GLUT1 expression is restored, which is paralleled by increased expression of vascular endothelial growth factor (VEGF) in macrophages at the BBB.
Obesity is associated with activation of a pro-inflammatory state in peripheral organs including white adipose tissue and liver leading to impairment of insulin action and glucose metabolism, ultimately resulting in development of type 2 diabetes mellitus (Odegaard and Chawla, 2013). However, the cause of increased inflammation in obesity is unknown. In preliminary experiments we have demonstrated that myeloid-cell derived VEGF is required to maintain brain glucose uptake only under conditions of high fat feeding in mice, indicating that activation of inflammation upon obesity serves as a homeostatic mechanism to reinstate glucose uptake to the brain. Reduction of brain glucose uptake appears to be a consequence of reduced expression of the glucose transporter (GLUT)-1, which is the major transporter of glucose in the CNS, where it is expressed in vascular endothelial cells of the blood-brain barrier (BBB) (Takata et al., 1997).
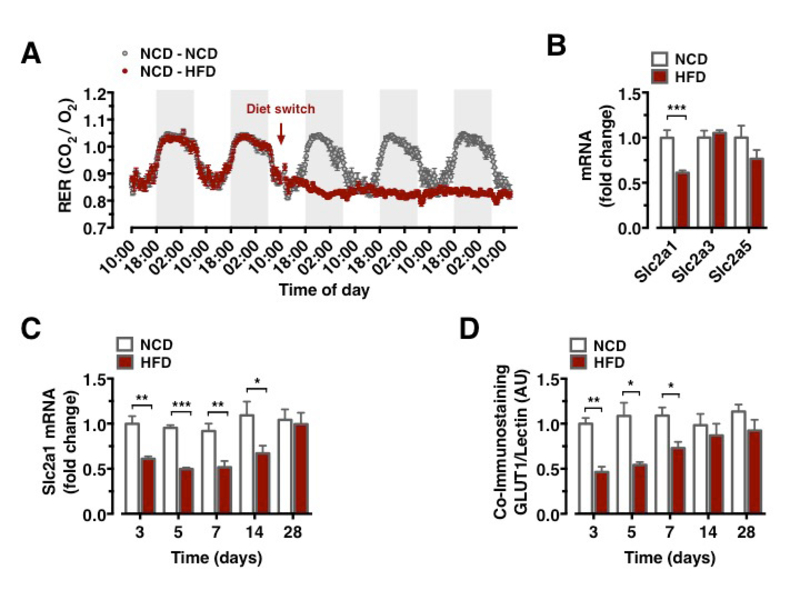
Metabolism during high-fat diet feeding acutely shifts from circadian, alternating carbohydrate and fat usage to a permanent predominance of fat utilization (Figure 1A). We compared the mRNA expression of critical glucose transporters in the CNS of mice, which had either remained on NCD feeding or had been switched to HFD feeding for 3 days. This analysis revealed a downregulation of Slc2a1 expression, while expression of Slc2a3 and Slc2a5 remained unaltered (Figure 1B). Brain Slc2a1 mRNA expression was transiently reduced as early as 3 days of HFD feeding and remained suppressed until 1 week of HFD feeding, while it was restored to comparable extend to that observed in constantly NCD fed animals after 4 weeks of HFD feeding (Figure 1C).
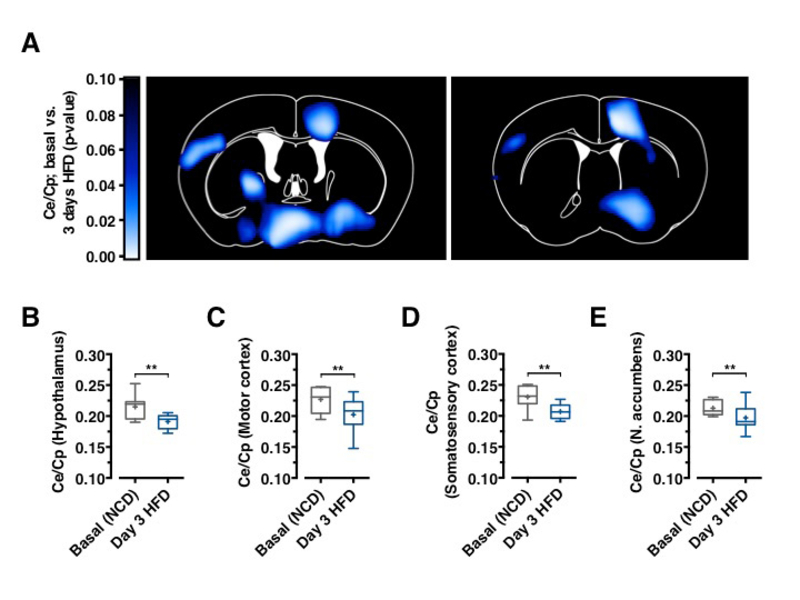
GLUT1 immunoreactivity was significantly reduced in BECs between 3 and 7 days after HFD feeding initiation (Figure 1D). Next, we aimed to directly investigate whether acute HFD feeding indeed reduced brain glucose uptake as suggested by reduced BBB-GLUT1 mRNA and protein expression. Further, we employed 18F-FDG positron emission tomography (PET) scans to quantitatively assess brain glucose uptake. Animals, which remained on NCD, expectedly revealed no decrease in brain glucose uptake (Data not shown), animals that were placed on HFD for 3 days exhibited significantly reduced glucose uptake in motor and sensory cortex, hypothalamus, and nucleus accumbens (Figures 2A–2E), revealing that diet-induced reduction of GLUT1 expression also functionally translated in reduced brain glucose uptake.
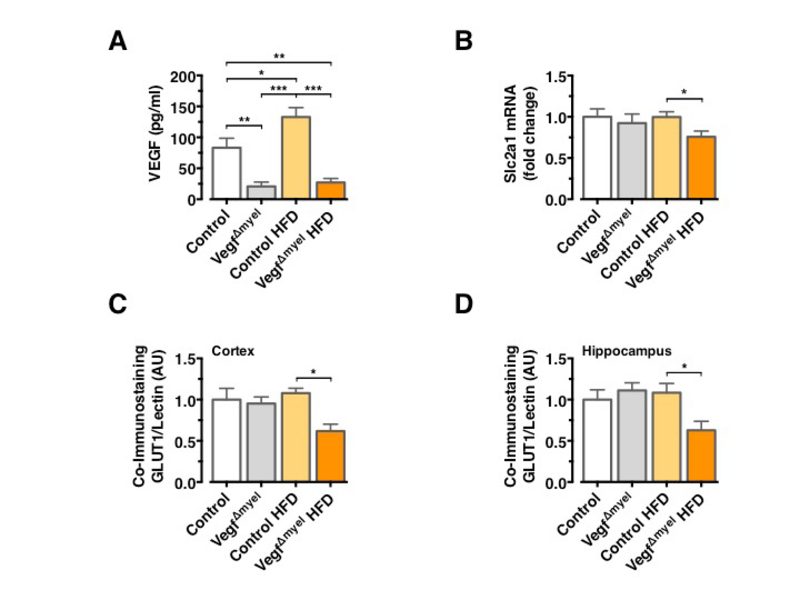
To further scrutinize the role of VEGF in control of brain glucose metabolism and systemic insulin action and resistance, we used mice with a specific Vegf deletion in cells of the myeloid lineage (VegfΔMyel). Analysis of serum VEGF levels confirmed the increase in circulating VEGF concentrations in control HFD-fed mice and revealed a profound reduction in circulating VEGF concentrations in VEGFΔmyel mice, both on NCD and HFD (Figure 3A). There was no significant difference in Slc2a1 mRNA expression between control and VEGFΔmyel mice fed an NCD (Figure 3B), indicating that myeloid-derived VEGF is not required to control BBB-GLUT1 expression in lean mice. However, VEGFΔmyel mice exhibited significantly reduced Slc2a1 mRNA expression compared to controls under HFD feeding (Figure 3B). GLUT1 immunoreactivity remained unaltered in control and VEGFΔmyel mice under NCD, while VEGFΔmyel mice exhibited significantly reduced GLUT1 immunoreactivity compared to controls under HFD (Figure 3C and 3D).
Consistent with the restoration of BEC GLUT1 expression 4 weeks after HFD feeding, in control animals prolonged HFD feeding did not result in reduced GLUT1 immunoreactivity in BECs, indicating that indeed upregulated VEGF can restore normal BBB-GLUT1 expression upon chronic HFD feeding even in long-term (Figure 3C and 3D).
These findings open conceptually a fundamental novel understanding of obesity-associated inflammation. Classically, obesity-associated inflammation has been viewed as a consequence of lipid overload and subsequent death of adipocytes, a process, which in turn recruits and activates macrophages to release pro-inflammatory cytokines. Given the critical role of tight control of brain glucose availability for organismal survival, activation of VEGF expression and inflammation upon transient reduction of brain glucose metabolism provides a prime mechanism to reinstate glucose availability to the CNS.
Odegaard and Chawla (2013). Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science vol. 339, pp. 172-177
Takata et al. (1997). Transport of glucose across the blood-tissue barriers. Int Rev Cytol vol. 172, pp. 1-53
Information from this funding period will not be updated anymore. New research related information is available here.

Policlinic for Endocrinology, Diabetes and Preventive Medicine
CMMC - PI - C 03
Executive Board Member
+49 221 478 32123
Policlinic for Endocrinology, Diabetes and Preventive Medicine
Kerpener Str. 62
50937 Cologne
Alexander Jais (PostDoc)
Maite Solas (PostDoc)
Sebastian Theurich (PostDoc)
Sophie Steculorum (PostDoc)
Brigitte Hampel (technician)
Julia Goldau (technician)
Jens Alber (technician)
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC