Lipid droplets (LD) are independent organelles composed of a neutral lipid core surrounded by a phospholipid monolayer. The function of LDs in neurons is totally obscure. Recently, functional studies of genes involved in hereditary spastic paraplegia (HSP) have suggested a putative role of LD disturbances in the pathogenesis of these conditions. Here we aim to tackle the functional link between LD metabolism and axonal degeneration.
Lipid droplets (LDs) are complex and dynamic organelles whose function is to assemble, store, and supply neutral lipids, mainly sterol esters and triacylglycerols (TAGs). Initially recognized in specialized cells, such as adipocytes, it is now clear that any cell has the ability to form LDs. Dysfunctions of LDs have been implicated in several pathologic conditions, such as obesity, atherosclerosis, and lipodystrophies. LDs are occasionally found in ultrastructural studies of neurons, however very little is known about their role in these cells. Notably, LDs appear to be increased in the brain of Alzheimer patients. Moreover, α-synuclein, the major constituent of Lewy bodies in Parkinson’s disease, was shown to accumulate on the surface of LDs in cells loaded with lipids. Recently, a link is emerging between LDs and axonopathies of the central nervous system, such as hereditary spastic paraplegia (HSP).
HSP is clinically defined by the association of weakness and spasticity of the lower limbs (pure HSP). The disease is caused by progressive retrograde degeneration of the longest axons of the central nervous system, those composing the corticospinal tract. HSP is one of the most heterogeneous neurological conditions from a genetic point of view. More than 80 genes have currently been identified to be responsible for this disease. A unifying view of HSP pathogenesis posits that alterations of the endoplasmic reticulum morphogenesis underlie this condition. Notably, several proteins implicated in HSP, such as Atlastin, REEP1, Seipin, Spartin, and DDHD2, have been linked to some aspect of LD function.
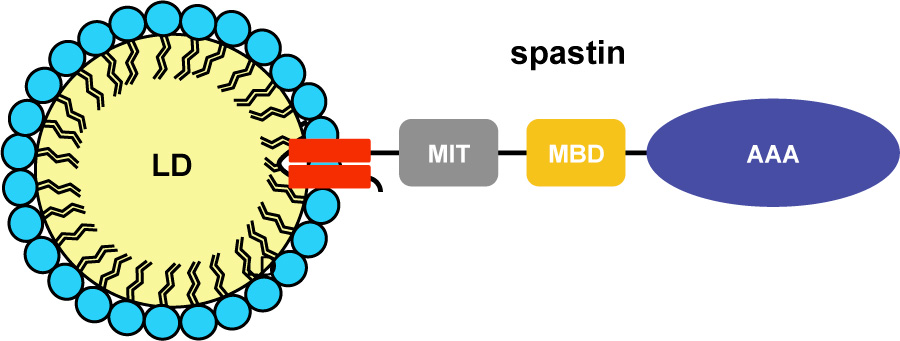
Our group has a long-standing interest in SPAST, encoding spastin, which is the most commonly mutated gene in patients affected by autosomal dominant hereditary spastic paraplegia (HSP). Spastin is a conserved microtubule (MT)-severing protein, involved in several processes requiring rearrangement of the cytoskeleton in concert to membrane remodeling, such as neurite branching, axonal growth, midbody abscission, and endosome tubulation. Two isoforms of spastin (spastin-M1 and spastin-M87) are synthesized from alternative initiation codons located in the first exon of the gene.
Recently, we found that in mammalian cells spastin-M1 can sort from the endoplasmic reticulum to pre- and mature lipid droplets (LDs) (Figure 1).
We further identified the N-terminal hydrophobic domain in cooperation with a basic motif in spastin-M1 to be necessary and sufficient for recruitment to LDs (Figure 2).
Increased levels of spastin-M1 expression lead to reduced number of bigger LDs, while expression of a spastin mutant unable to bind and sever MTs causes clustering of LDs. The role of spastin in LD formation and lipid metabolism is evolutionary conserved.
Downregulation of Dspastin and expression of a dominant-negative variant decrease LD number in Drosophila nerves and skeletal muscle, while Dspastin overexpression leads to an opposite phenotype. Ubiquitous downregulation or upregulation of Dspastin levels affects triacylglycerol levels in a consistent fashion.
Moreover, we found reduced amount of fat stores in intestinal cells of worms in which the spas-1 homologue was either depleted by RNA interference or deleted. Taken together, our data uncovers a role of spastin in LD metabolism and strongly suggest that dysfunction of LDs in axons may contribute to the pathogenesis of HSP.
We have unravelled a link between spastin and lipid droplet metabolism. However, several questions remain to be addressed. What is the function of spastin at LDs in mammalian cells? Is the LD phenotype a primary effect of spastin deficiency? Is a disturbance in LD metabolism relevant in axonal degeneration? Is the microtubule severing activity of spastin implicated in regulating LD biogenesis?
Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jungst C, Tzschichholz A, Schauss A, Honing S, Trifunovic A, Daga A, and Rugarli EI (2015). Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet 11, e1005149.
Information from this funding period will not be updated anymore. New research related information is available here.

CECAD Cologne & Institute for Genetics
CMMC - PI - C 14
show more…+49 221 478 84244
+49 221 478 97902
CECAD Cologne & Institute for Genetics
Joseph-Stelzmann-Str. 26
50931 Cologne
David Pla-Martin (PostDoc)
Desiree Schatton (PhD student)
Simon Schumacher (PhD student)
Nimesha Tadepalle (PhD student)
Sara Murru (PhD student)
Marie-Charlotte Marx (PhD student)
Esther Barth (Technician)

Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC