We are focussed on the role of cell death in health and disease. In particular, we investigate the molecular mechanisms regulating different forms of cell death (e.g. apoptosis and necroptosis) and how cell death contributes to inflammatory programs, activated following pathogenic infection or tissue damage, as well as to tumorigenesis. Understanding the complex relationship between cell death and inflammation will help to improve the current treatments of chronic inflammatory diseases and cancer.
Programmed cell death is a fundamental biological process that ensures tissue development and homeostasis. However, when deregulated, cell death can induce inflammation. Genetic studies conducted in mice demonstrated that aberrant cell death is a causative factor for chronic inflammatory syndromes.
In the past few years it became clear that the cell death processes causing different autoinflammatory diseases are mediated by a serine/threonine kinase called RIPK1. Indeed, pharmacological inhibition of RIPK1 has been showed to block cell death and ameliorate inflammatory conditions such as psoriasis and inflammatory bowel disease.
It has also been shown that RIPK1-mediated cell death of cancer cells has the potential to alert and activate the immune system to mount an anti-tumour immune response. Therefore, while inhibition of RIPK1 activity appeared to be beneficial in auotinflammatory diseases, boosting its cytotoxic potential might be useful in conditions where the activation of the immune system against cancer cells is the desired outcome.
One of our focuses is on how cell death is regulated downstream of different innate immune receptors, such as TNFR1. Once activated by TNF, TNFR1 initiates a cascade of events that trigger the activation of NF-kappaB and MAPKs, leading to the expression of pro-survival and pro-inflammatory genes (Figure 1). However, under certain circumstances, TNFR1 activation can also induce cell death in the form of apoptosis or necroptosis. Apoptosis is mediated by the kinase activity of RIPK1 and the proteolytic activity of Caspase-8, necroptosis is mediated by the kinase activity of RIPK1 and RIPK3 and the pseudokinase MLKL (Figure 1). The main goal of our work is to characterise novel mechanisms determining whether TNF stimulation induces the expression of pro-inflammatory genes or cell death and to understand the impact of these two different outcomes on tissue plasticity.
Characterizing how the life and death decision is made downstream of TNFR1 will provide a better understanding of how the immune responses are regulated followed pathogen infection or tissue injury.
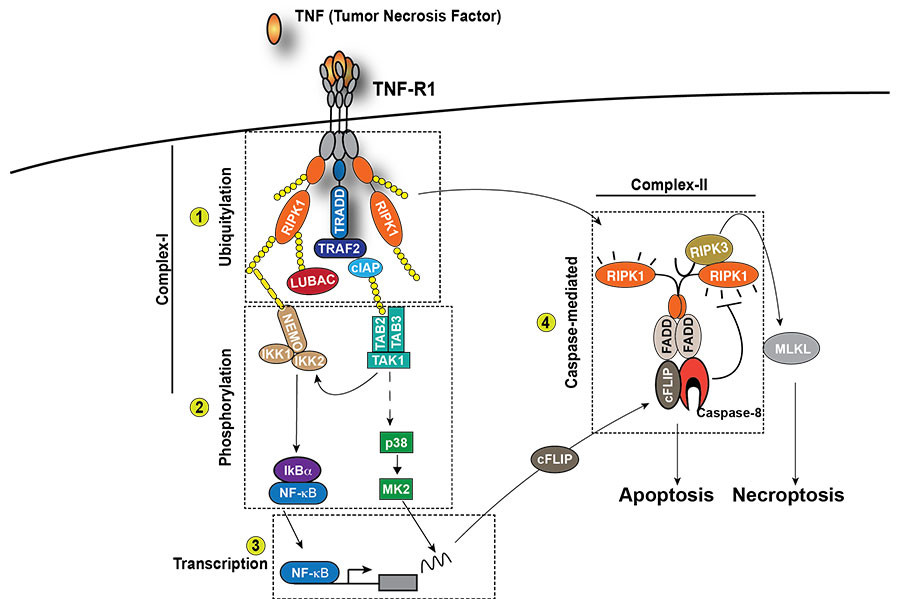
Apoptosis and necroptosis are the best understood forms of programmed cell death. It was generally thought that apoptosis is immunologically silent, since dying cells maintain plasma membrane integrity. In contrast, necroptosis is an immunogenic form of cell death as a consequence of plasma membrane disruption and spillage of existing immune-stimulatory cellular factors referred to as constitutive Damage Associated Molecular Patterns (cDAMPs). This traditional viewpoint has recently been challenged. It becomes increasingly clear that the type of cell death per se (apoptosis vs. necroptosis or other forms of programmed cell death) does not determine whether dying cells trigger effector T cell (CD8+)-mediated immune responses. CD8+T cell activation indeed requires not only malignant cell death as source of antigens, but also the active production and release of immune-stimulatory factors, called inducible DAMPs (iDAMPs) by dying cells. This ensures robust T cell cross priming and activation against malignant cells (Figure 2).
A recent study indicated RIPK1 can induce immunogenic cell death in malignant cells, regardless of the type of cell death delivered (apoptosis or necroptosis) but based on the ability of RIPK1 to activate an inflammatory program in dying cells. The mechanisms regulating RIPK1 activity have been extensively characterised in pathophysiological settings. However, the signals upstream and downstream of RIPK1 that determine its ability to induce immunogenic cell death in malignant cells are unknown. The characterisation of such signals might help the development of novel strategies to maximise the activation of the immune system against cancer cells and improve current immunotherapy.
It is widely accepted that cell death does not only regulate organism development and the elimination of unwanted cells, but it also actively participates to tissue repair programs. Such programs are activated following damage or infection and are also often hijacked by cancer cells to fuel their own expansion. Characterising the mechanisms by which cell death is regulated and the role of cell death in tissue repair and adaptation will help the understanding of the processes underlying autoinflammatory diseases and tumorigenesis. Equally, the characterisation of how cell death can be exploited to activate anti-tumour immune responses might ultimately help to improve current anti-cancer therapies.
For further information please check here the Annibaldi Laboratory For Cell Death, Inflamation and Immunity's webpage.
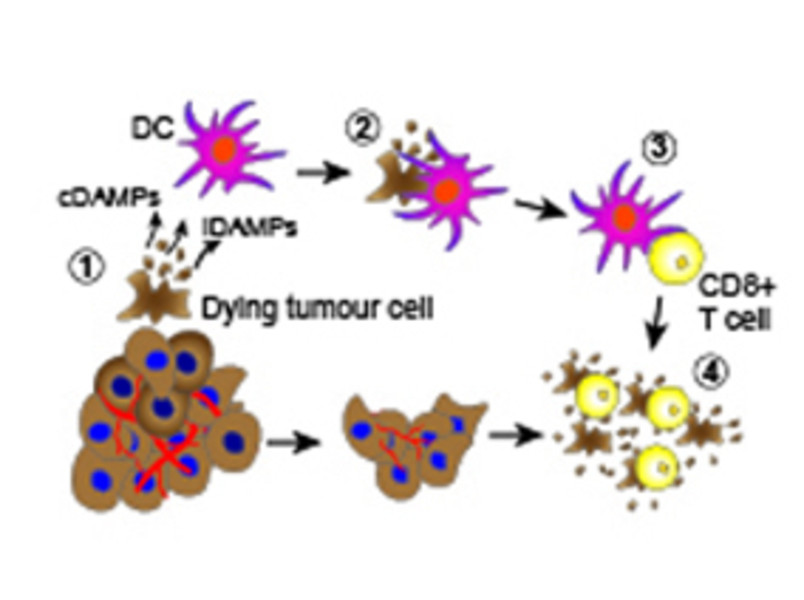
Dying cancer cells release immune-stimulatory factors (cDAMPs and iDAMPs) that recruit dendritic cell (DC) (1), which, in turn, phagocytose the dying cancer cells (2) and present tumor antigens to CD8+ T cells (cross-presentation) (3) determining their activation (cross-priming). Activated CD8+ T cells selectively recognize and eliminate tumor cells (4) contributing to tumor eradication.

Center for Molecular Medicine Cologne | Lab. of Cell Death, Inflammation and Immunity - CMMC Research Building
CMMC - Co-PI - A 09
CMMC - PI - JRG 12
alessandro.annibaldi[at]uk-koeln.de
show more…+49 221 478 37455
Center for Molecular Medicine Cologne | Lab. of Cell Death, Inflammation and Immunity - CMMC Research Building
Robert-Koch-Str. 21
50931 Cologne
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC