We generated an unbiased mammalian model, the fads2-/- mouse, deprived of ω3- and ω6-PUFA synthesis. The fads2-/- mouse is auxotrophic and an ideal experimental platform for deciphering the molecular basis underlying the role of PUFAs in cell biology, membrane and nutritional research and altered lipid homeostasis in related pathologies. Fads2 is an antagonistic pleiotropic gene.
Central to intense research efforts for several decades has been the systemic role of ω3- and ω6-polyunsaturated fatty acids (PUFAs) as constituents of membrane phospholipid (PL) bilayers and in lipid homeostasis and related imbalances in several pathologies of dyslipidemias, and as driving force in the development of chronic metabolic, vascular and neurodegenerative disorders. Many of these disorders have been proposed as therapeutic targets of ω3-PUFA (docosahexaenoic acid) substituted diets, despite a molecular understanding of their function is lacking.
We overcame the lack of a mammalian model by generation of the ω3- and ω6-PUFA deprived fads2-/- in vivo mouse model, which allowed a constructive experimental basis for the molecular approach in deciphering the role of PUFAs in cell biology, membrane- and nutritional research and altered lipid homeostasis in related pathologies.
A novel 20:35,11,14- acid (sciadonic acid), so far not observed in mammalian tissues, is synthesized from the essential linoleic acid in the fads2-/- mutant. 20:35,11,14 is incorporated as an arachidonic acid surrogate.
The nd-fads2-/- mutant is auxotrophic, an essential prerequisite for unbiased dietary studies. Supplementation of arachidonic acid (AA, ω6-20:4) and docosahexaenoic acid (DHA, ω3-22:6) to the normal diet transform the nd-fads2-/- into the „AA-fads2-/-“ with 20:4 and „DHA-fads2-/-“ with 22:6 as single PUFA–substituent in all phospholipids.
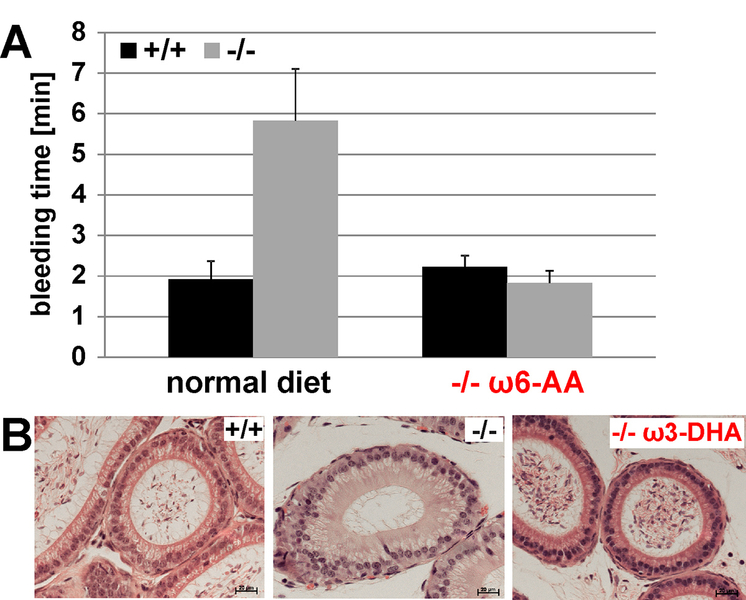
The major phenotypes of the fads2-/- mice are infertility and disturbed hemostasis. Both can be overcome by supplementation of DHA or AA respectively (figure1).
Arachidonic acid has been discovered in 1990 by R. Mechuoulam as precursor of endocannbinoids N-arachidonoylethanolamide (anandamide; AEA) and sn-2-arachidonoylglycerol (2-AG), which are ligands of endocannabinoid receptors CB1 and 2, and substrates of fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase, forming the mammalian Endocannabinoid system (ECS). Endocannabinoids exert their biological effects by means of binding to G protein-coupled receptors (GPCR). The CB1 receptor is found predominantly in brain, with highest densities in hypothalamus, hippocampus, cerebellum and striatum. The CB2 receptor is found mainly in spleen and hematopoietic cells. Sources of arachidonic acid are a) the essential fatty acid linoleic acid being Δ6-desaturated, chain elongated and Δ5-desaturated, and b) diet. The biosynthetic pathways of a) AEA from N-arachidonoyl-phoshatidylethanolamide (NAPE) by phospholipase D hydrolysis and b) 2-AG released by DAG-lipase from diacylglycerol (DAG), the DAG backbone of PIPs, are well established.
We will analyze the ECS and the metabolic, cognitive and physical role in the PUFA-deficient nd-fads2-/- and the AA-fads2-/- and DHA-fads2-/- mice.
The most but also controversially discussed issue is the role of PUFAs in close correlation to lipoprotein pattern and development of atherosclerotic plaque formation. The two canonical mouse models of rodent atherosclerosis, the apoE-/- and ldlr-/- mouse mutants, have been valuable tools in atherosclerosis research. These mutants rapidly develop massive plaques in the aortic arc and the abdominal aorta. We crossed the fads2-/- gene locus into these two mutants, which yielded the double mutants fads2-/-x apoE-/- and fads2-/- x ldlr-/- for feeding experiments with standard “Western diet” (WD).
The PUFA-pattern in these double mutants can be manipulated by controlled dietary regimens. These ideal unbiased genetic models will pave the way not only to assess or disprove the widely accepted view of the beneficial role of PUFAs in the prevention of atherosclerosis but also provide insight into the underlying molecular pathology.
Studies on the lipoprotein (Lp) metabolism correlated with the pathogenesis of plaque formation in the apoe-/- and ldlr-/- mouse mutants.
The severely altered lipid bilayers of subcellular membranes in the fads2-/- liver and the rapid response of the auxotrophic mutant to AA- and DHA- supplemented dietary regimens (Stoffel et al. 2014) suggested studies on serum lipoprotein synthesis, assembly and secretion and directed the focus to the role of ω3- and ω6-PUFAs in dyslipidemias and associated atherosclerosis?”
Preliminary feeding experiment of the two double mutants with WD gave encouraging results, which indicate a protective role of the deleted fads2-locus, as indicated in the oil red stained aorta assembly presented in figure2.
The fads2-/- mouse lends itself as a powerful tool to prove the role of ω6- and ω3-PUFAs as benefactor or culprit on the molecular, cellular and systemic level. In dietary studies the mouse serves as a genetically unbiased model for the titration of ω6- and ω3-PUFAs and thus of the most important currently known epigenetic factor.
Stoffel, W., Holz, B., Jenke, B., Binczek, E., Guenter, R., Kiss, C., Karakesisoglou, I., Thevis, M., Weber, A., Arnhold, S., and Addicks, K. (2008). Δ6-Desaturase (FADS2) deficiency unveils the role of ω3- and ω6-polyunsaturated fatty acids. The EMBO Journal 27, 2281–2292.
Stoffel, W., Hammels, I., Jenke, B., Binczek, E., Schmidt-Soltau, E., Brodesser, S., Odenthal, M., and Thevis, M. (2014). Obesity resistance in Δ6- fatty acid desaturase (FADS2) deficiency. EMBOR 25, 110-120.
Hammels, I., Binczek, E., Schmidt-Soltau, I., Jenke, B., Thomas, A., Vogel, M., Thevis, M., Filipova, D., Papadopoulos, S., and Stoffel, W. (2019). Novel CB1-ligands maintain homeostasis of the endocannabinoid system in omega3- and omega6-long-chain-PUFA deficiency. J Lipid Res 60, 1396-409.
Stoffel, W., Hammels, I., Jenke, B., Schmidt-Soltau, I., and Niehoff, A. (2019). Neutral Sphingomyelinase 2 (SMPD3) Deficiency in Mice Causes Chondrodysplasia with Unimpaired Skeletal Mineralization. Am J Pathol 189, 1831-45.
Stoffel W, Jenke B, Schmidt-Soltau I, Binczek E, Brodesser S, and Hammels I (2018). SMPD3 deficiency perturbs neuronal proteostasis and causes progressive cognitive impairment. Cell Death Dis 9, 507.
Stoffel W, Schmidt-Soltau I, Jenke B, Binczek E, and Hammels I (2017). Hair Growth Cycle Is Arrested in SCD1 Deficiency by Impaired Wnt3a-Palmitoleoylation and Retrieved by the Artificial Lipid Barrier. J Invest Dermatol 10.1016/j.jid.2017.02.973.
Information from this funding period will not be updated anymore. New research related information is available here.
Antagonistic pleiotropy of the Δ6-fatty acid desaturase (fads2) gene in disease and senescence. ω3- and ω6-Polyunsaturated fatty acids in Δ6-fatty acid desaturase (fads2-/-) deficiency

Laboratory of Molecular Neuroscience - Center for Biochemistry
CMMC - PI - SRG 01
Executive Board Member
wilhelm.stoffel[at]uni-koeln.de
show more…+49 221 478 6881
+49 221 478 6882
Laboratory of Molecular Neuroscience - Center for Biochemistry
Joseph-Stelzmann-Str. 52
50931 Cologne
Dr. rer. nat. Ina Hammels (PostDoc)
Erika Binczek (technician)
Britta Jenke (technician)
Inga Schmidt-Soltau (technician)
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC