Bronchopulmonary dysplasia (BPD), a neonatal chronic lung disease, is characterised by inflammation and lung growth arrest. The lack of therapies emphasizes the need to define new therapeutical strategies. We aim to decipher the mechanistic function of Klf4 – a regulator of cell pluripotency and inflammation – in regenerative and inflammatory processes. Identification of Klf4 as a novel target to promote alveolarization could define new avenues to enable growth and regeneration of lungs with BPD.
Preterm infants, whose lungs are incompletely developed, are at high risk forrespiratory failure. Oxygen (O2) and mechanical ventilation (MV) offer life-saving treatments for respiratory distress; such treatment, however, leads to bronchopulmonary dysplasia (BPD), a neonatal chronic lung disease. Despite the advances in perinatal care, BPD is the most common complication of prematurity. Lungs of infants afflicted with BPD are characterized by impaired formation of alveoli and pulmonary microvessels as well as perturbed matrix remodelling, yielding structural changes resembling emphysema or chronic obstructive lung disease (COPD). The high incidence of BPD and the lack of therapies emphasize the need of unravelling the early molecular origins that cause the initial lung injury to offer novel targets promoting lung growth. To this end, we used two animal models of neonatal lung growth arrest, hyperoxia- and ventilation-induced lung injury. Interestingly, we found that Krüppel-like factor 4 (klf4), a zinc-finger transcription factor that plays a major role in regulating cell pluripotency, cell survival and cell differentiation, is markedly reduced in neonatal lungs with growth arrest. Based on our previous findings we aim to define the functional role of Klf4 in lung regenerative mechanisms to enable lung growth after neonatal lung injury.
The alveolar epithelium is composed of alveolar epithelial type I cells (ATI) and ATII. Over 95 % of the alveolar surface is covered by ATI, representing the gas exchange surface; whereas ATII secrete surfactant proteins to reduce surface tension, and are immunologically active. Progress in stem cell biology demonstrates progenitor properties of ATII. Understanding how ATII allows alveolar growth or regeneration is clinically relevant to treat chronic lung diseases. Since Klf4 regulates cell pluripotency and is essential in cell differentiation, we studied Klf4 in ATII during normal and aberrant lung development using prolonged O2or lengthy MV. First, Klf4 expression was markedly up-regulated and localized with ATII during normal alveolarization. Conversely, reduced Klf4 was related to loss of ATII in our animal models of lung growth arrest. Complimentary cell culture studies demonstrated that oxygen-induced inhibition of cell survival of lung epithelial cells was linked to reduction of Klf4 and impaired cell homeostasis, suggesting that Klf4 regulates epithelial-mesenchymal transition (EMT) in ATII.
Further analyses using overexpression and CRISPR/Cas9-mediated deletion identified Klf4 as a key regulator of lung epithelial cell homeostasis viaa novel Smad2-Stat3 protein interaction. Moreover, translational studies showed that ATII-specific ablation of Klf4 during alveolarization promoted ATII differentiation and enabled lung growth. These findings highlight Klf4 as a novel target to promote lung growth and regeneration.
Secondary septation is a process during late lung development, which comprises normal subdivision of primitive air sacs into mature alveoli. Myofibroblasts are central for this process by producing lung matrix and secreting growth factors. Our group recently showed that aberrant lung growth with perturbed matrix is linked to dysfunction of myofibroblasts. Since Klf4 is involved in fibrotic process, ongoing studies from our group are currently deciphering the mechanistic role of Klf4 in primary neonatal lung fibroblasts (pnF). We linked a loss of Klf4 in neonatal lungs to increased number of myofibroblasts after prolonged O2. Similarly, exposure of pnF to O2blunted Klf4 expression. Further in vitrostudies identified Klf4 to be crucial in migration, proliferation, apoptosis and expression of matrix molecules of pnF. Investigation of the Klf4 interactome using BioID provides novel insight in the regulatory network of Klf4 and new targets to preserve physiological fibroblast function and possibly treat fibrosis.
The pathogenesis of BPD includes inflammation, but the mechanisms are not yet known. Macrophages are key constituents of lung inflammation, secreting various cytokines, such as Interleukin 6 (IL6), CXCL10 and MMP12, regulatorof cell homeostasis, matrix remodelling, immune response and tissue regeneration.Interestingly,Klf4 has been identified as a key regulator of macrophage differentiation, maintaining anti-inflammatory M2 phenotype; in contrast, deletion of Klf4 favours inflammatory M1. Infants evolving BPD, show an elevation of M1 markers, whereas M2 markers are unchanged or even reduced. Similarly, in models of neonatal lung growth arrest a macrophage influx is linked to increased protease activity and macrophage-related cytokines, e.g. IL6. We next tested if blockade of these cytokines enables lung formation after hyperoxia. Indeed, IL6 and MMP12 null mice were partly protected from hyperoxia-induced lung injury. In particular, loss of IL6 preserved survival and homeostasis of ATII, alveolar formation, and ultimately lung function, suggesting IL6 as a novel therapeutical target to treat BPD.
BPD remains a devastating disease of extreme premature infants. Our future perspective aims to target Klf4 in three major pathomechanisms of BPD: (1) regenerative capacity; (2) matrix remodelling; and (3) inflammation. Effective treatment or prevention of hyperoxia- or ventilator-induced lung injury evolving into BPD likely will derive from elucidating molecular mechanisms causing initial injury, which is the central goal of our research group.
Mohr J, et al., IL-6/Smad2 signaling mediates acute kidney injury and regeneration in a murine model of neonatal hyperoxia. FASEB J. 2019.
Nawabi J, et al.,Novel functional role of GH/IGF-I in neonatal lung myofibroblasts and in rat lung growth after intrauterine growth restriction. Am J Physiol Lung Cell Mol Physiol. 2018.
Alejandre Alcazar MA et al., Elafin treatment rescues EGFR-Klf4 signaling and lung cell survival in ventilated newborn mice. Am J Respir Cell Mol Biol. 2018.
Will, J.P., Hirani, D., Thielen, F., Klein, F., Vohlen, C., Dinger, K., Dotsch, J., and Alcazar, M.A.A. (2019). Strain-dependent effects on lung structure, matrix remodeling, and Stat3/Smad2 signaling in C57BL/6N and C57BL/6J mice after neonatal hyperoxia. American Journal of Physiology-Regulatory Integrative and Comparative Physiology 317, R169-R81.
Alcazar MAA, Kaschwich M, Ertsey R, Preuss S, Milla C, Mujahid S, Masumi J, Khan S, Mokres LM, Tian L, Mohr J, Hirani DV, Rabinovitch M, and Bland RD (2018). Elafin Treatment Rescues EGFR-Klf4 Signaling and Lung Cell Survival in Ventilated Newborn Mice. American Journal of Respiratory Cell and Molecular Biology 59, 623-634.
Ferrari N, Bae-Gartz I, Bauer C, Janoschek R, Koxholt I, Mahabir E, Appel S, Alcazar MAA, Grossmann N, Vohlen C, Brockmeier K, Dotsch J, Hucklenbruch-Rother E, and Graf C (2018). Exercise during pregnancy and its impact on mothers and offspring in humans and mice. Journal of Developmental Origins of Health and Disease 9, 63-76.
Nawabi J, Vohlen C, Dinger K, Thangaratnarajah C, Klaudt C, Garcia EL, Hirani DV, Karakaya PH, Macheleidt I, Odenthal M, Nusken KD, Dotsch J, and Alcazar MAA (2018). Novel functional role of GH/IGF-I in neonatal lung myofibroblasts and in rat lung growth after intrauterine growth restriction. American Journal of Physiology-Lung Cellular and Molecular Physiology 315, L623-L637.
Information from this funding period will not be updated anymore. New research related information is available here.
Netrin-1 as a new target to treat chronic lung disease: Identification of agonists and antagonists by artificial intelligence
Pursuing novel molecular mechanisms and treatment strategies in bronchopulmonary dysplasia: functional role of Krüppel-like factor 4 (Klf4)

Clinic for Pediatric and Adolescent Medicine
CMMC - PI - assoc. 38
CMMC - former PI - CAP 10
miguel.alejandre-alcazar[at]uk-koeln.de
show more…+49 221 478 96876
+49 221 478 32400
Clinic for Pediatric and Adolescent Medicine
Kerpener Str. 62
50937 Cologne
Dharmesh Hirani (doctoral student)
Jasmine Mohr (Postdoc)
Christina Vohlen (technician).

Figure 1: Schematic model depicting the role of Krüppel-like factor 4 (Klf4) in lung growth after exposure to increased oxygen or mechanical ventilation.
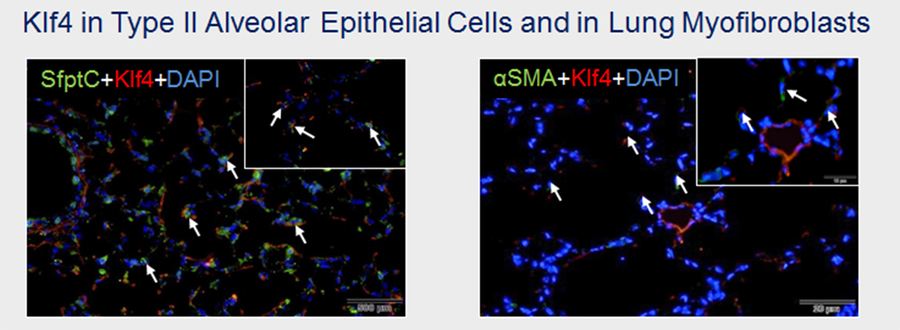
Figure 2: Co-localization of Klf4 with type II alveolar epithelial cells (Surfactant protein C; SfptC) and myofibroblasts (αSMA), in lungs of newborn mice.
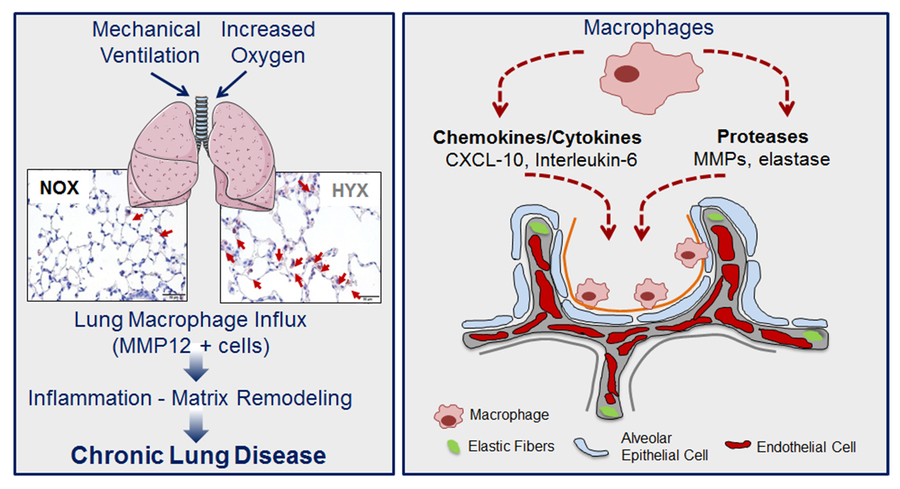
Figure 3: Schematic model depicting the impact of increased oxygen and mechanical ventilation on lung inflammation; normoxia, NOX; hyperoxia; HYX.
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC