Cell death and inflammation are common features of many autoimmune and degenerative disorders. It is now clear that cell death can trigger chronic inflammation resulting in autoimmunity and degenerative diseases, including cancer. Therefore, understanding the crosstalk between death and inflammation is important to find effective therapies for such diseases. We aim to investigate the processes regulating the execution of cell death downstream of innate immune receptors. In particular, we want to uncover the importance of different modalities of cell death in autoimmune diseases and cancer, with the ultimate purpose to find to therapeutic strategies to tackle inflammation-dependent pathologies.
Cell death is crucial to maintain tissue homeostasis as it is important for tissue repair and infectious processes. Consequently, many organisms can sense both damage and pathogens, a deed achieved via the so-called damage- and pathogen-associated molecular patterns (DAMPs and PAMPs, respectively) that are recognised by Pattern-recognition receptors (PRRs). The PRR family consists of Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and DNA sensors such as DNA-induced activator of interferon (IFN) (DAI, also known as ZBP1) and cyclic GMP-AMP synthase (cGAS). Triggering of PRRs results in the induction of inflammatory cytokines and chemokines including, among others, TNF, IL-1 and type I IFNs. These cytokines play crucial roles in triggering innate immune responses by binding to their respective receptors, including TNF receptor (TNFR) superfamily (SF) members.
Binding of TNF to its cognate receptor TNFR1 results in downstream cascades that induce: i) inflammation/survival via LUBAC and cIAP1/2, by activating NF-B/MAPK-mediated gene expression, ii) apoptosis via FADD/RIPK1/caspase-8 or, iii) necroptosis via RIPK1 autophosphorylation, RIPK3 and MLKL (Fig. 1). In normal physiology, TNFR1-signalling output is skewed towards inflammation/survival; however, in autoimmune disorders or pathological conditions this balance is shifted towards cell death induction.
In the past years, we discovered that The Linear Ubiquitin chain Assembly Complex (LUBAC), an E3 ligase that generates linear ubiquitin chains on components of many immune receptor-signaling complexes, is crucial for survival as it prevents both apoptosis and necroptosis (Fig. 2A). In a model of LUBAC deficiency specifically in the skin, which results in severe dermatitis due to excessive cell death (Fig. 2B), we further found that combined loss of several DRs (namely TNFR1, CD95 (Fas) and TRAIL-R), but not their individual loss, prevented chronic inflammation. Thus, we identified CD95 and TRAIL-R, in addition to TNFR1, as important DRs in cell death-induced inflammation, providing support for combining DR-inhibition as treatment of inflammation-associated autoimmunity.
The general concept is that necroptosis is highly immunogenic, due to the release of immunogenic factors such as DAMPs, whilst apoptosis is immunologically silent. However, this concept has been challenged and in certain circumstances apoptosis can also trigger immunogenicity. It is not only the mode of cell death itself to dictate immunogenicity, but also the inflammatory pathways that are concomitantly activated, and the consequent release of immunogenic factors. The decision between survival and death and the cell death programs that are activated will determine a pathophysiological outcome. Therefore, it is important to define which and how cell death processes shape immune responses in health and disease.
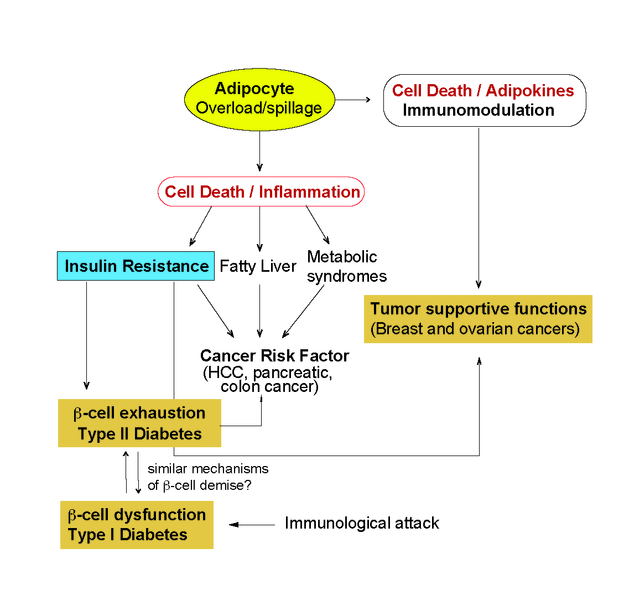
One common autoimmune disease with a cell death signature is Type 1 Diabetes (TID), which is caused by an immune reaction against pancreatic β-cells. However, the exact mechanisms dictating β-cell demise responsible for activating immune responses during T1D are currently poorly understood. We focus on understanding the signaling cascades that mediate cell death and inflammation in T1D and in identifying β-cell death mechanisms that induce or perpetuate immunogenicity. Diabetes is also induced by obesity. Yet, in this case, it is called Type II Diabetes (TIID) and the specific immune attack of β-cells is less clear. Instead, obesity and the associated insulin resistance ultimately lead to β-cell exhaustion which reduces β-cell mass. Again, the mechanisms of β-cell demise in TIID are not entirely clear. We want to study the mechanisms dictating β-cell death in the context of autoimmunity and obesity and find common and unique features in TI/IID.
Chronic, low-grade inflammation of adipose tissue plays a key role in the pathogenesis of obesity. Both adipocytes and the macrophages that accumulate in adipose tissue during obesity are source of inflammatory cytokines and have been identified as key components in the development of chronic inflammation and consequent insulin resistance (Fig. 3). Obesity-associated inflammation is a prominent cause of cancer. Increasing adiposity leads to adipocyte death and accumulation of macrophages in the so-called crown-like structures (CLS) which has been shown to have tumor supportive functions. The type of adipocyte death that dictates the formation of tumor supportive CLS as well as the molecular mechanisms leading to recruitment of adipocyte associated macrophages are still not completely understood. We propose to study the interplay between cell death and obesity-induced inflammation and understand the contribution of adipocyte death during obesity in the tumor microenvironment.
SCLC is an aggressive type of solid tumor that represents approx. 15% of lung cancer cases. Whereas chemotherapy typically elicits high initial response rates, it is followed by disease recurrence and fatal progression. Dinaciclib is a CDK9 inhibitor that is currently in phase-III clinical testing. CDK9i results in downregulation of the anti-apoptotic factors cFLIP, cIAP1/2 and MCL-1. In addition, Dinaciclib-treated tumor cells express and release factors that are typical of immunogenic cell death (ICD). We therefore predict that boosting immunogenicity by CDK9 inhibition could synergize with chemotherapy. We plan to examine how CDK9 inhibition modulates cell death in SCLC and how can this be exploited to overcome resistance in combination with chemo- and/or immunotherapy.
We aim to uncover which and how cell death pathways modulate disease outcome. We aim to build a research programme in which basic and translational research are closely aligned so as to be able to translate fundamental discoveries on the mechanism and aetiology of disease into novel treatments options for patients.
Information from this funding period will not be updated anymore. New research related information is available here.
Cell death and inflammation in health and disease

Dept. of Translational Genomics & Center for Molecular Medicine Cologne - CECAD Research Center
CMMC - PI - assoc. JRG 03 and CAP 24
show more…
+49 221 478 84083
Dept. of Translational Genomics & Center for Molecular Medicine Cologne - CECAD Research Center
Joseph-Stelzmann Strasse 26
50931 Cologne


Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC