Motoneurons (MNs) are highly polarized nondividing cells that innervate muscles via the neuromuscular junctions, the largest synapses in our body. MNs must be maintained throughout life otherwise neuropathies and muscle atrophy will develop. During development, external cues - such as netrins - direct the axons to their ultimate destination by interacting with specific attractive or repulsive neuronal receptors. To accomplish this unique feature, axons and dendrites rely on highly sophisticated cellular processes - such as efficient local protein translation - that enable a prompt response to internal signals and external cues. In MNs, local protein translation requires a proper transport of mRNAs and cargos, a coordinated autophagy and protein degradation system, and functional mitochondria to provide enough energy locally for all these processes.
In this project we will make use of mouse and cellular models of Spinal Muscular Atrophy (SMA) -the most common recessively inherited MN disorder in humans - to understand why reduced levels of the house-keeping SMN protein primarily led to dysfunction of MNs. In addition, we will take advantage of our knowledge about three SMA protective modifiers (PLS3, NCALD and CHP1), which can counteract SMA development in humans and various SMA species by restoring impaired endocytosis (Ref 1-14). Nevertheless, how endocytosis is affected in SMA MNs and how it ultimately affects MN function, particularly axonal local translation, mitochondrial bioenergetics and neurotransmission, is still poorly understood and it will be the main objective of this project.
In particular, we will focus on these 3 specific questions:
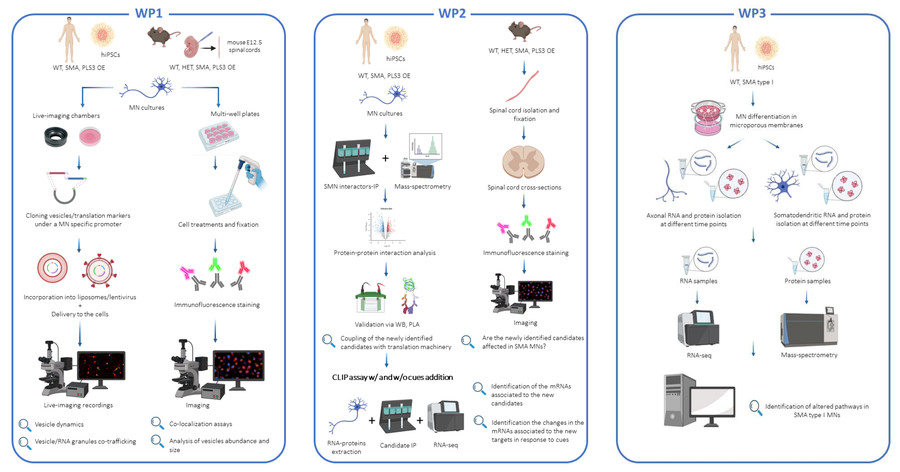
Axonal vesicle trafficking and local translation are key regulatory mechanisms affected in a plethora of MN disorders, including SMA. In SMA, these processes are understudied, especially in the axons and pre-synapses. Recently, three FDA- and EMA-approved therapies, aimed at increasing the levels of functional SMN protein, showed impressive improvement in SMA patients. However, the current therapies are not efficient for severely affected SMA patients, who require very high levels of SMN protein postnatal, and for adult SMA patients, who require only low levels of SMN protein. The identification of new molecular mechanisms that can support MN function is needed to develop new therapies to reduce MN alterations and counteract their loss in SMA patients. Here, we aim to investigate these mechanisms and contribute to the development of SMN-independent therapies for SMA and MN disorders in general.
In this project we will use the following cutting-edge approaches (Figure 1) that will enable us to elucidate outstanding questions in the field of vesicle-trafficking, axonal translation and selective vulnerability in healthy and SMA motor neurons.


Institute of Human Genetics - CMMC Research Building
CMMC - PI - C 18
CMMC - Executive Board Member
brunhilde.wirth[at]uk-koeln.de
show more…+49 221 478 86464
+49 221 478 86465
Institute of Human Genetics - CMMC Research Building
Kerpener Str. 34
50931 Cologne
https://humangenetik.uk-koeln.de/forschung/ag-neuromuskulaere-und-skelettale-erkrankungen/

Institute of Human Genetics - CMMC Research Building
CMMC - Co-PI - C 18
JRG Leader - Institute for Human Genetics Cologne
valentina.piano[at]uk-koeln.de
show more…+49 221 478 89523
Institute of Human Genetics - CMMC Research Building
Robert-Koch-Str. 21
50931 Cologne
PostDoc
Mert Karakaya
PhD students
Eleonora Zilio
Salim Caglar Avci
Nur Cengiz-Winter
Ilka Maus
Tamas Schmidt
Jorge Arturo Soriano Campos
Sofia Vrettou
Lisa Wolff
Medical student
Jaqueline Lenz
Technician
Irmgard Hölker
Roman Rombo
Sebastian Zetzsche
Britta Meiger
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC