Polycystic kidney diseases (PKD) affect millions of people worldwide and are the major genetic cause of kidney replacement therapy (KRT) in both children and adults. The two major types of PKD are the more common autosomal-dominant polycystic kidney disease (ADPKD), mainly caused by variants in the PKD genes PKD1 and PKD2, and the rare pediatric autosomal-recessive polycystic kidney disease (ARPKD), mainly caused by PKHD1 variants. Notably, there is clinical and genetic overlap between ARPKD and ADPKD. All three genes encode for proteins localized to primary cilia. Cilia are sensory organelles that in the kidney project from the apical membrane of epithelial cells into the lumen of the tubules. PKD is generally regarded as a paradigm ciliopathy. In contrast to ADPKD, for which a first disease-modifying pharmacological therapy has been established, there is currently no targeted therapy for ARPKD.
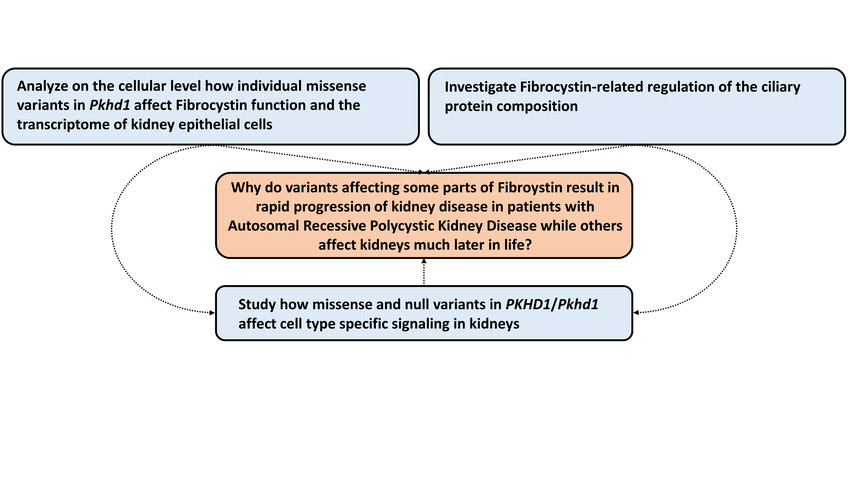
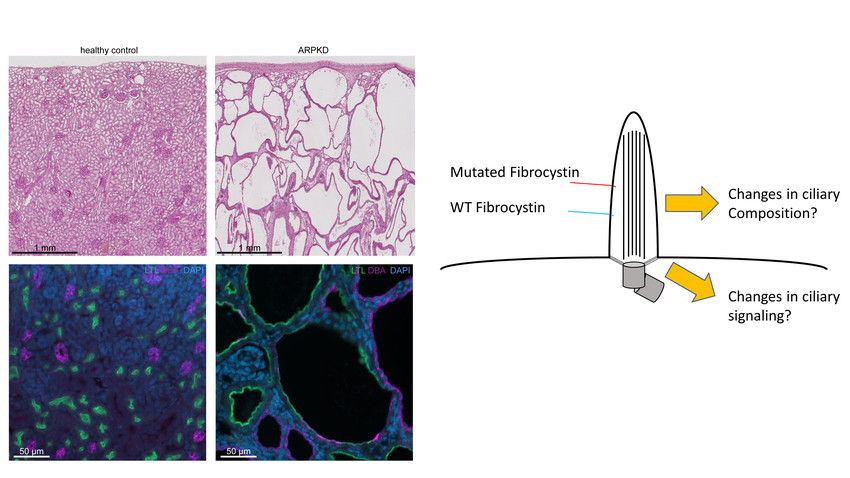
PKHD1 encodes for the large ciliary transmembrane protein fibrocystin (FC). Disease-causing variants in PKHD1 have been described almost across the entire gene. By analyzing longitudinal clinical data of hundreds of ARPKD patients in a large multinational collaborative effort, we could recently gain significant insights into the genotype-phenotype correlation of ARPKD patients. We could show that missense variants in specific areas of the gene are associated with better kidney survival. With the ARegPKD registry initiated by one of the applicants, we have full access to the largest clinical ARPKD database worldwide. Moreover, we could generate and confirm the recently described first Pkhd1-associated preclinical model of ARPKD that results in a kidney and liver phenotype recapitulating human ARPKD. Using a translational approach by combining our novel insights into genotype-phenotype correlations with available with the novel mammalian in vivo model, we follow the overarching aim to uncover the mechanisms of how variants in Pkhd1 result in PKD by affecting primary cilia and transcriptional programs in kidney cells. In particular, we aim to understand why variants affecting some parts of FC result in rapid progression of kidney disease while others affect kidneys much later in life. Specifically, we will (1) analyze on the cellular level how individual missense variants in Pkhd1 affect FC function and the transcriptome of kidney epithelial cells, (2) investigate FC-related regulation of the ciliary protein composition in vitro and in vivo, and (3) study how missense and null variants in PKHD1/Pkhd1 affect cell type specific signaling in kidneys. Ultimately, we aim to contribute to the development of novel disease-modifying therapeutic concepts for ARPKD.
Autosomal-recessive polycystic kidney disease (ARPKD) is a severe and early-onset pediatric disease characterized by fibrocystic changes in kidneys and the liver. It is a major cause of chronic kidney disease and kidney failure in children requiring dialysis or transplantation. There is no targeted therapy yet as the molecular pathogenesis is not well understood. Here, we aim to combine the power of clinical knowledge obtained from a pan-European registry study with state-of-the-art preclinical models, cellular biochemical approaches, and single-cell transcriptomics to identify the relevant pathways that distinguish between early and late renal involvement. We anticipate that our work will result in a deeper disease understanding paving the way for strategies to delay the onset and progression of kidney disease in ARPKD and additional renal ciliopathies.
Introduction
Clinical Relevance

Clinic and Polyclinic for Pediatric and Adolescent Medicine
CMMC - PI - C 11
show more…+49 221 478 4359
+49 221 478 97295
Clinic and Polyclinic for Pediatric and Adolescent Medicine
Kerpener Straße 62
50937 Cologne
Department of Pediatrics
Center for Family Health
Center for Rare Diseases Cologne

Clinic II of Internal Medicine
CMMC - Co-PI - C 11
bernhard.schermer[at]uk-koeln.de
show more…+49 221 478 89030
+49 221 478 6360
Clinic II of Internal Medicine
CECAD Research Building, Joseph-Stelzmann-Str. 26
50931 Cologne
PostDoc
Claudia Dafinger
Kathrin Burgmaier
Jopp Hillen
Büsra Yildirim
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC