Spinal muscular atrophy (SMA) is caused by homozygous absence of the survival motor neuron gene 1(SMN1) and inability of SMN2to compensate for the SMN1loss. We found that increased levels of plastin 3 (PLS3) and reduced levels of neurocalcin delta (NCALD) protect against SMA in humans and across species (zebrafish, worm and mice). However, the molecular genetic cause for the modified expression of these modifiers in asymptomatic SMN1-deleted individuals is not understood, but relevant for future therapies. Moreover, these protective modifiers helped us to uncover – 20 years after gene discovery- impaired endocytosis as a main cellular mechanism disturbed in SMA; the exact underlying mechanisms is unknown.
The motor neuron (MN) disease spinal muscular atrophy (SMA) is caused by reduced levels of the survival of motor neuron (SMN) protein, whose function is critical for assembly of the spliceosome and other protein complexes. Despite the housekeeping function, SMN deficit in SMA patients primarily affects the function of spinal MN and especially of the neuromuscular junction (NMJ), resulting in dysfunction, degeneration and consequent muscular atrophy.
We identified two human-protective SMA modifiers - PLS3 upreg-ulation and NCALD downregulation - and proved their ability to counteract SMA in vitroand in vivousing SMA zebrafish and mouse models. Both proteins and their interactome helped us to uncover endocytosis as a major cellular pathway disturbed in SMA and rescued by these modifiers. Here, we address the following two questions:
We found that Plastin 3 (PLS3) is upregulated in genetically predisposed but phenotypically asymptomatic women, and acts protective against SMA (Oprea et al 2008). The protective effect of PLS3 upregulation has been confirmed in various genetically-modified SMA models (worm, fly, zebrafish and mice) as well as by gene therapy using AAV9-PLS3 (Oprea et al 2008; Ackermann et al 2013; Hosseinibarkooie et al 2016; Kaifer et al 2017).
PLS3is upregulated in approximately 5% of the healthy population, while the reason is still elusive. As PLS3is localized on the X-chromosome, we hypothesised that PLS3might escape the X-chromosomal inactivation (XCI), since no other variants within PLS3 were identified that may explain this unusual finding (Oprea et al 2008).
Using a multi-OMICS approach, including genome sequencing, transcriptome (RNASeq) from various cell lines (lymphoblastoid cell lines, fibroblasts and iPSC-derived motor neurons), epigenetic analysis as well as proteome analysis, we aim to unveil the underlying cis and trans-regulatory network leading to the cell-type specific PLS3 upregulation. We found that PLS3 in asymptomatic individuals is upregulated in lymphoblastoid cell lines and MN-derived from iPCSs, but not in the fibroblast cell line from which these originated. This suggest a cell type-specific regulation. Transcriptome and SNP analyses of single cell clones of iPSC-derived MNs showed biallelic expression of PLS3but not of most other X-linked genes in asymptomatic but not SMA individuals, indicating that PLS3 escapes XCI in asymptomatic individuals. PLS3is located adjacent to the unique macrosatellite DXZ4. This macrosatellite has a highly variable number of a 3 kb repeat monomer in humans, and is essential for XCI. Indeed, DXZ4is associated with heterochromatic epigenetic features on the active X-chromosomes, while it has euchromatic features on the inactive X-chromosomes and the locus of DXZ4 is therefore open for transcription. We hypothesised that the repeat number of DXZ4modulates the expression of PLS3and other adjacent genes on the inactivated X-chromosome facilitating their escape from XCI. To determine the repeat number of DXZ4, we established a novel technique called molecular combing and compared women that differ in their PLS3expression. This method uses multi-colour DNA probes to mark repeat regions on linear stretched DNA molecules.
By this, we quantified the repeat number of DXZ4in SMA affected and asymptomatic women and compared the results to PLS3 expression levels measured by qRT-PCR. Asymptomatic women with a high PLS3 expression showed at least one allele with a high copy number repeat as compared to the symptomatic one with none or low PLS3 levels. Currently, we perform 4C-sequencing to identify the regulatory landscape of PLS3and DXZ4. Moreover, we are currently establishing a methylation sensitive assay to verify the skewed XCI. Overall, this is a highly challenging, but promising and exciting project, for which a DFG-funding will be submitted soon.
NCALD shows a Ca2+-dependent interaction with clathrin, an important protein, coating synaptic vesicles during endocytosis. We found that NCALD binds clathrin only when the Ca2+level is low. Using patch clamp and Ca2+imaging, we demonstrate that the Ca2+influx is reduced in Smn-depleted PC12 cells. However, this was not restored by simultaneous NCALD reduction. Thus, we assumed that another mechanism must be responsible for the positive effect of NCALD reduction on endocytosis. Indeed, we show that NCALD binds clathrin only in the presence of low calcium levels, as is the case in SMA cells. Hence, a reduced amount of NCALD releases clathrin, resulting in an increased endocytosis rate.
Moreover, under stimulation of different cell types as well as ex vivoin NMJs, endocytosis was strongly impaired in SMA, whereas NCALD reduction restored it (Riessland et al 2017). Moreover, we identified calcineurin-like EF-hand protein1 (CHP1) as a third SMA protective modifier (Janzen et al 2018). In NSC34 cells, Chp1knockdown tripled macro-pinocytosis whereas clathrin-mediated endocytosis remained unaffected. Importantly, Chp1knockdown restored macropinocytosis in Smn-depleted cells by elevating calcineurin phosphatase activity. CHP1 is an inhibitor of calcineurin, which collectively dephosphorylates proteins involved in endocytosis, and is therefore crucial in synaptic vesicle endocytosis. Indeed, we found marked hyperphos-phorylation of dynamin1 in SMA motor neurons, which was restored to control level by the heterozygous Chp1mutant allele (Janzen et al 2018).
In collaboration with IONIS Pharmaceuticals we test antisense oligonucleotides (ASOs) against Ncaldand Chp1to study the power of combinatorial therapy in SMA.
Upadhyay A, … Wirth B.Neurocalcin Delta Knockout Impairs Adult Neurogenesis Whereas Half Reduction Is Not Pathological. Front Mol Neurosci.2019;Feb 12;12:19. doi: 10.3389/fnmol.2019.00019.
Janzen, E., … Wirth B.CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain2018, 141 (8), 2343-2361
Mendoza-Ferreira N, … Wirth B.Biallelic CHP1 mutation causes human autosomal recessive ataxia by impairing NHE1 function. Neurol Genet2018, 4: e209
Riessland M*, Kaczmarek A*, Schneider S*, ... Wirth B.Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet,2017; 2. Feb. 100 (2): 297-315
Patent: B. Wirth,E. Janzen, N. Mendoza-Ferreira, SM Hosseinibarkooie, EP 17172826, Calcineurin B Homologous Protein 1 Inhibitors and Therapeutic and Non-Therapeutic Uses Thereof,4filed May. 24, 2017
Karakaya, M., Paketci, C., Altmueller, J., Thiele, H., Hoelker, I., Yis, U., and Wirth, B. (2019). Biallelic variant in AGTPBP1 causes infantile lower motor neuron degeneration and cerebellar atrophy. Am J Med Genet A 179, 1580-4.
Schorling, D.C., Becker, J., Pechmann, A., Langer, T., Wirth, B., and Kirschner, J. (2019). Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology 93, 267-9.
Upadhyay, A., Hosseinibarkooie, S., Schneider, S., Kaczmarek, A., Torres-Benito, L., Mendoza-Ferreira, N., Overhoff, M., Rombo, R., Grysko, V., Kye, M.J., Kononenko, N.L., and Wirth, B. (2019). Neurocalcin Delta Knockout Impairs Adult Neurogenesis Whereas Half Reduction Is Not Pathological. Front Mol Neurosci 12, 19.
Torres-Benito, L., Schneider, S., Rombo, R., Ling, K.K., Grysko, V., Upadhyay, A., Kononenko, N.L., Rigo, F., Bennett, C.F., and Wirth, B. (2019). NCALD Antisense Oligonucleotide Therapy in Addition to Nusinersen further Ameliorates Spinal Muscular Atrophy in Mice. Am J Hum Genet 105, 221-30.
Diao Y, Cui L, Chen Y, Burbridge TJ, Han W, Wirth B, Sestan N, Crair MC, and Zhang J (2018). Reciprocal Connections Between Cortex and Thalamus Contribute to Retinal Axon Targeting to Dorsal Lateral Geniculate Nucleus. Cereb Cortex 28, 1168-1182.
Ghosh SG, Becker K, Huang H, Dixon-Salazar T, Chai G, Salpietro V, Al-Gazali L, Waisfisz Q, Wang H, Vaux KK, Stanley V, Manole A, Akpulat U, Weiss MM, Efthymiou S, Hanna MG, Minetti C, Striano P, Pisciotta L, De Grandis E, Altmuller J, Nurnberg P, Thiele H, Yis U, Okur TD, Polat AI, Amiri N, Doosti M, Karimani EG, Toosi MB, Haddad G, Karakaya M, Wirth B, van Hagen JM, Wolf NI, Maroofian R, Houlden H, Cirak S, and Gleeson JG (2018). Biallelic Mutations in ADPRHL2, Encoding ADP-Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome. Am J Hum Genet 103, 431-439.
Goncalves I, Brecht J, Thelen MP, Rehorst WA, Peters M, Lee HJ, Motameny S, Torres-Benito L, Ebrahimi-Fakhari D, Kononenko NL, Altmuller J, Vilchez D, Sahin M, Wirth B, and Kye MJ (2018). Neuronal activity regulates DROSHA via autophagy in spinal muscular atrophy. Sci Rep 8, 7907.
Janzen E, Mendoza-Ferreira N, Hosseinibarkooie S, Schneider S, Hupperich K, Tschanz T, Grysko V, Riessland M, Hammerschmidt M, Rigo F, Bennett CF, Kye MJ, Torres-Benito L, and Wirth B (2018). CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 10.1093/brain/awy167.
Karakaya M, Storbeck M, Strathmann EA, Vedove AD, Holker I, Altmueller J, Naghiyeva L, Schmitz-Steinkruger L, Vezyroglou K, Motameny S, Alawbathani S, Thiele H, Polat AI, Okur D, Boostani R, Karimiani EG, Wunderlich G, Ardicli D, Topaloglu H, Kirschner J, Schrank B, Maroofian R, Magnusson O, Yis U, Nurnberg P, Heller R, and Wirth B (2018). Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum Mutat 10.1002/humu.23560.
Krosschell KJ, Kissel JT, Townsend EL, Simeone SD, Zhang RZ, Reyna SP, Crawford TO, Schroth MK, Acsadi G, Kishnani PS, Von Kleist-Retzow JC, Hero B, D'Anjou G, Smith EC, Elsheikh B, Simard LR, Prior TW, Scott CB, Lasalle B, Sakonju A, Wirth B, Swoboda KJ, and Project Cure SMAIsN (2018). Clinical trial of L-Carnitine and valproic acid in spinal muscular atrophy type I. Muscle Nerve 57, 193-199.
Martinez Carrera LA, Gabriel E, Donohoe CD, Holker I, Mariappan A, Storbeck M, Uhlirova M, Gopalakrishnan J, and Wirth B (2018). Novel insights into SMALED2: BICD2 mutations increase microtubule stability and cause defects in axonal and NMJ development. Hum Mol Genet 27, 1772-1784.
Mendoza-Ferreira N, Coutelier M, Janzen E, Hosseinibarkooie S, Lohr H, Schneider S, Milbradt J, Karakaya M, Riessland M, Pichlo C, Torres-Benito L, Singleton A, Zuchner S, Brice A, Durr A, Hammerschmidt M, Stevanin G, and Wirth B (2018). Biallelic CHP1 mutation causes human autosomal recessive ataxia by impairing NHE1 function. Neurol Genet 4, e209.
Neugebauer J, Heilig J, Hosseinibarkooie S, Ross BC, Mendoza-Ferreira N, Nolte F, Peters M, Holker I, Hupperich K, Tschanz T, Grysko V, Zaucke F, Niehoff A, and Wirth B (2018). Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum Mol Genet 10.1093/hmg/ddy318.
Shorrock HK, van der Hoorn D, Boyd PJ, Llavero Hurtado M, Lamont DJ, Wirth B, Sleigh JN, Schiavo G, Wishart TM, Groen EJN, and Gillingwater TH (2018). UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain 141, 2878-2894.
Strathmann EA, Peters M, Hosseinibarkooie S, Rigo FW, Bennett CF, Zaworski PG, Chen KS, Nothnagel M, and Wirth B (2018). Evaluation of potential effects of Plastin 3 overexpression and low-dose SMN-antisense oligonucleotides on putative biomarkers in spinal muscular atrophy mice. PLoS One 13, e0203398.
Diao Y, Cui L, Chen Y, Burbridge TJ, Han W, Wirth B, Sestan N, Crair MC, and Zhang J (2017). Reciprocal Connections Between Cortex and Thalamus Contribute to Retinal Axon Targeting to Dorsal Lateral Geniculate Nucleus. Cereb Cortex10.1093/cercor/bhx028, 1-15.
Hosseinibarkooie S, Schneider S, and Wirth B (2017). Advances in understanding the role of disease-associated proteins in spinal muscular atrophy. Expert Rev Proteomics 14, 581-92.
Karakaya M, Mazaheri N, Polat I, Bharucha-Goebel D, Donkervoort S, Maroofian R, Shariati G, Hoelker I, Monaghan K, Winchester S, Zori R, Galehdari H, Bonnemann CG, Yis U, and Wirth B (2017a). Biallelic MCM3AP mutations cause Charcot-Marie-Tooth neuropathy with variable clinical presentation. Brain 140, e65.
Karakaya M, Yilmaz S, Storbeck M, Hoelker I, Heller R, Serdaroglu G, Gokben S, Yis U, and Wirth B (2017b). PRUNE1: a disease-causing gene for secondary microcephaly. Brain 140, e61.
Krosschell KJ, Kissel JT, Townsend EL, Simeone SD, Zhang RZ, Reyna SP, Crawford TO, Schroth MK, Acsadi G, Kishnani PS, Von Kleist-Retzow JC, Hero B, D'Anjou G, Smith EC, Elsheikh B, Simard LR, Prior TW, Scott CB, Lasalle B, Sakonju A, Wirth B, Swoboda KJ, and Project Cure SMAIsN (2017). Clinical trial of L-carnitine and valproic acid in spinal muscular atrophy type I. Muscle Nerve10.1002/mus.25776.
Neri M, Scotton C, Gualandi F, Wirth B, Schols L, Klockgether T, Lochmuller H, Muntoni F, D'amico A, Bertini E, Pane M, Mercuri E, and Ferlini A (2017). Genetic landscapes in neuromuscular disorders: The influence of next-generation sequencing analysis. European Journal of Neurology 24, 500-.
Rademacher S, Verheijen BM, Hensel N, Peters M, Bora G, Brandes G, Vieira de Sa R, Heidrich N, Fischer S, Brinkmann H, van der Pol WL, Wirth B, Pasterkamp RJ, and Claus P (2017). Metalloprotease-mediated cleavage of PlexinD1 and its sequestration to actin rods in the motoneuron disease spinal muscular atrophy (SMA). Hum Mol Genet 26, 3946-59.
Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Lohr H, Bradler C, Grysko V, Dimitriadi M, Hosseinibarkooie S, Torres-Benito L, Peters M, Upadhyay A, Biglari N, Krober S, Holker I, Garbes L, Gilissen C, Hoischen A, Nurnberg G, Nurnberg P, Walter M, Rigo F, Bennett CF, Kye MJ, Hart AC, Hammerschmidt M, Kloppenburg P, and Wirth B (2017). Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am J Hum Genet 100, 297-315.
Salpietro V, Lin W, Vedove AD, Storbeck M, Liu Y, Efthymiou S, Manole A, Wiethoff S, Ye Q, Saggar A, McElreavey K, Krishnakumar SS, Group SS, Pitt M, Bello OD, Rothman JE, Basel-Vanagaite L, Hubshman MW, Aharoni S, Manzur AY, Wirth B, and Houlden H (2017). Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann Neurol 81, 597-603.
Storbeck M, Horsberg Eriksen B, Unger A, Holker I, Aukrust I, Martinez-Carrera LA, Linke WA, Ferbert A, Heller R, Vorgerd M, Houge G, and Wirth B (2017). Phenotypic extremes of BICD2-opathies: from lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. Eur J Hum Genet 25, 1040-8.
Information from this funding period will not be updated anymore. New research related information is available here.
Targeting CHP1, an SMN-independent pathway to counteract spinal muscular atrophy in mice and human iPSCs

Institute of Human Genetics - CMMC Research Building
CMMC - PI - C 18
CMMC - Executive Board Member
brunhilde.wirth[at]uk-koeln.de
show more…+49 221 478 86464
+49 221 478 86465
Institute of Human Genetics - CMMC Research Building
Kerpener Str. 34
50931 Cologne
https://humangenetik.uk-koeln.de/forschung/ag-neuromuskulaere-und-skelettale-erkrankungen/
Mohsenseyyed Hosseinibarkkoie (PostDoc)
Eva Janzen (PostDoc
Natalia Mendoza-Ferreira (PostDoc)
Svenja Schneider (Postdoc)
Laura Torres-Benito (PostDoc)
Andrea delle Vedove (MD/PhD student)
Mert Karakaya (MD student)
Anixa Muinos Brühl (PhD student)
Ilka Müller (PhD student)
Bryony Ross (MD/PhD student)
Tamas Schmidt (PhD student)
Eike Strathmann (PhD student)
Aaradhita Upaydhyay (PhD student)
Charlotte Veltman (MD student
Lisa Wolff (Master student)
Charlotte Dresen (Bachelor student)
Michelle Scharte (Bachelor student)
Irmgard Hölker (TA)
Kristina Hupperich (TA)
Roman Rombo
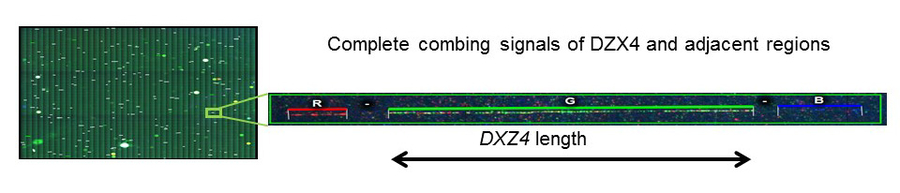
Figure 1:Visualization of DZX4by molecular combing
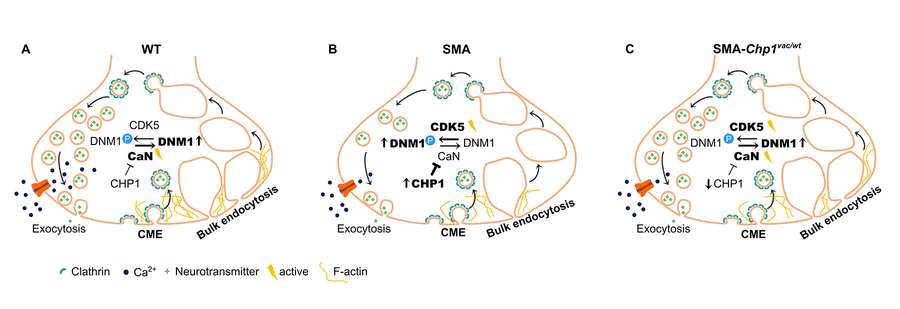
Figure 2: CHP1downregulation improves endocytosis by increasing calcineurinphosphatase activity (Janzen et al 2018).
Copyright ©
Prof. Dr. med. Thomas Benzing
Chair of the CMMC